



Brain reserve is an important concept to understand the variability of damage associated with brain-related diseases and includes the adaptation of cognitive processes to preserve brain function. A good cognitive reserve might delay the onset of clinical manifestations of neurodegenerative diseases as well as hepatic encephalopathy, improving the quality of life in patients with chronic liver diseases. By stimulating activities and maintaining overall health, individuals may be able to enhance their brain's resilience to age-related changes and pathology. This review aims to collect all the data available on the role of brain reserve in hepatic encephalopathy development, and the potential effect of a good brain reserve in slowing down hepatic encephalopathy progression and frequency.
Brain reserve is a term used to describe the brainʼs ability to protect itself against the onset of cognitive impairment and brain-related diseases. Brain reserve includes two concepts which are cognitive reserve and brain maintenance. Cognitive reserve includes the adaptation of cognitive processes for age, diseases, and insults. In cases where a disease results in brain damage, the brain processes assist in adjusting and preserving compensatory cognitive functions [1]. Hepatic encephalopathy is a critical clinical complication in patients with acute and chronic liver failure. The pathophysiology involves multiple mechanisms, but the main one is mediated by the accumulation of ammonium, a neurotoxin that damages cognitive reserve [2].
2Pathophysiological mechanisms in hepatic encephalopathy2.1The role of ammonium accumulation, mitochondrial disruption, and neuronal deathCognitive impairment in patients with liver diseases is attributed to ammonium accumulation, reduced brain glycogen levels, neurotransmitter disruption, and neuroinflammation. Active and passive components cause it. The passive component refers to the threshold for resisting brain aging, such as brain size. The active component is a process to improve the effect of brain aging, for example, the ability to recruit compensatory neural structures to replace damaged pathways, these pathways include education and occupation therapy [3]. Poor calory intake damages brain reserve disrupting proteins located in cortical structures. It is a requirement for regeneration of this tissue which improves with an adequate caloric intake. Deep slow-wave sleep has been demonstrated to enhance cognitive function [4].
Hepatic encephalopathy is caused by many cerebral pathways that increase or decrease brain reserve. Glycogen is a polysaccharide of glucose that serves as a form of energy storage in the liver and skeletal muscle. However, the brain has low glycogen storage, which is expressed in neuronal and glial cells, mainly located in astrocytic areas with a higher concentration in high synaptic density areas involving neural activity [5,6].
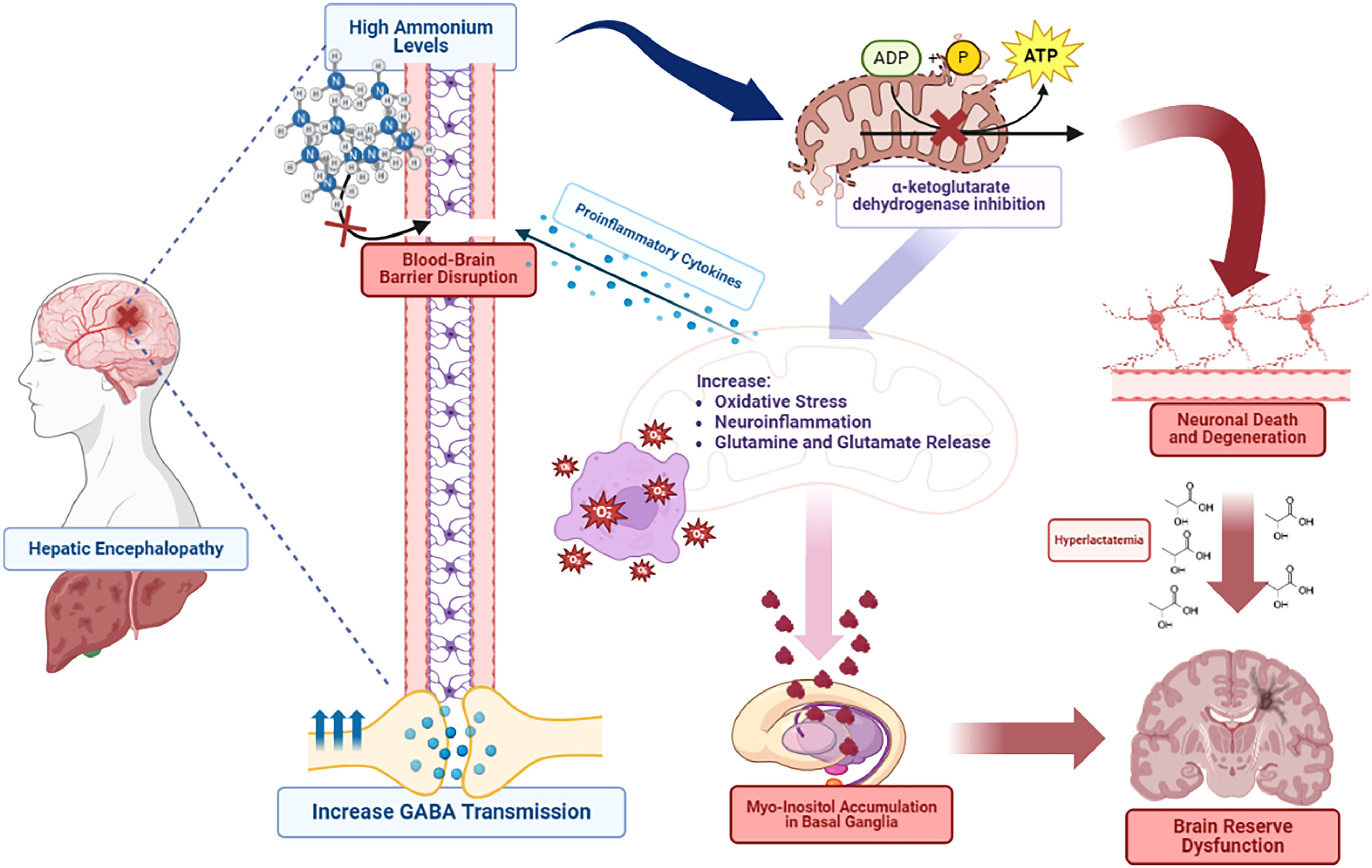
There is evidence that complications of a high concentration of ammonium can lead to altered intracellular pH, increased inhibitory neurotransmission, and cerebral edema. An enzyme known as alpha-ketoglutarate dehydrogenase is inhibited by high ammonium levels, disrupting mitochondrial metabolism and energy production of neuronal cells. Gamma-aminobutyric acid (GABA) participates in inhibitory neurostimulation and is believed to be increased secondary to high glutamine concentrations. On the other hand, hyperammonemia activates the opening of the blood-brain barrier leading to making the brain more susceptible to endotoxemia by cyclooxygenase mechanisms [7,8].
Hepatic encephalopathy leads to mitochondrial dysfunction and neuronal death. The central nervous system (CNS) depends on ATP production by mitochondria. Damage through oxidative phosphorylation causes an ATP disruption and altered energy disposition in neurons [9]. It is known that ammonium inhibits tricarboxylic acid cycle (TCA) enzymes, and inhibits mitochondrial permeability, increases oxidative stress, neuroinflammation, and glutaminergic transmission (Fig. 1). Another important mechanism of cognitive dysfunction is astrocytic senescence, through alteration of signal growth factors and synaptic glutamate homeostasis. Senescence participates in many enzymatic pathways, which causes an increase in free iron levels in cells. In addition, it contributes to the activation of p21 and p53 pathways to decrease the survival of neuronal cells [10].
, and myo-inositol accumulation in basal ganglia which leads to neuronal death and brain reserve dysfunction.")
Hepatic Encephalopathy is associated with hyperammonemia. It disrupts the blood-brain barrier and mitochondrial damage, by inhibiting α-ketoglutarate dehydrogenase. Consequently, it causes neuronal degeneration and an increase in oxidate stress, neuroinflammation, and glutamine release. Glutamine excess leads to stimulation of GABA neurotransmitters, proinflammatory cytokines (IL-1, IL6, TNF-a), and myo-inositol accumulation in basal ganglia which leads to neuronal death and brain reserve dysfunction.
Some studies found that hepatic encephalopathy patients had a significant reduction in cerebral perfusion and oxygen metabolic rate. When episodes of encephalopathy resolved, these levels improved, and patients had a significant decrease in astrocytes’ oxidative metabolism [11,12]. Hyperammonemia alters cerebral oxygen levels, contributing to the downregulation of endothelial nitric oxide synthase. Nitric oxide plays a vital role in controlling microvascular perfusion causing low oxygen levels and increasing lactate production, which disrupts cognitive reserve [13].
Glucose metabolism is altered in hepatic encephalopathy patients. Consequently, metabolic pathways are directed to alternative pathways, leading to lactate production. Accumulation of this substrate is known to contribute to brain metabolic alterations and elevated intracranial pressure [9]. There is an association between hyperammonemia, and inhibition of α-ketoglutarate dehydrogenate which stimulates glutamate dehydrogenase, activating alternative pathways for lactate production. Hyperlactatemia results in cerebral osmotic stress in addition to increased water formation due to oxidate phosphorylation. Glutamine production disrupts mitochondrial function, opening transition pores which cause astrocytes’ edema [14]. Microglia secrete proinflammatory cytokines and activate NLRP3 triggering brain dysfunction. Furthermore, this systemic and cerebral hypersecretion of proinflammatory cytokines induces activation of NFkB leading to blood–brain barrier weakening [15].
Ammonium alters dopaminergic and serotonergic neurotransmitter pathways, releasing glutamine and glutamate by astrocytes. It causes overstimulation of N-methyl-aspartate receptors leading to neuromodulation, neurodegeneration, and neural apoptosis. All this is through intracellular hypercalcemia that reduces the neuronal activity of the nitric oxide GMP pathway, damaging neurotransmission. Patients with cognitive dysfunction present an increase of myo-inositol in the thalamus, putamen, and white matter, decreasing the glutamine/glutamate ratio [10]. However, a reduction in dopamine levels was seen in the hippocampus with a high D1 and D2 receptor expression. Consequently, it perpetuates brain damage and creases brain resilience, disrupting cognitive reserve [16].
Hepatic encephalopathy damages brain energy and protein metabolism. Stimulation of glycolysis increases the production of lactate and glutamine, causing the mobilization of amino acids. It provides a carbonyl group as a substrate for TCA for the detoxification of ammonium through the synthesis of glutamine, increasing cerebral proteolysis [17]. Animal studies demonstrate that liver cirrhosis generates neurometabolic changes in the hippocampus by the increase in glutamine concentration. Manganese level elevation and brain deposits in astrocytic cells are an important mechanism of hepatic encephalopathy. This is because cholestasis slows down manganese excretion, leading to frontal cerebral atrophy, astrocytic mitochondrial toxicity, and high oxidative stress, which contributes to inflammation and cerebral edema [18]. In addition, high manganese levels interfere with the glutamine-glutamate cycle, affecting neuronal metabolism and neurotransmission, which results in memory impairment and cognitive dysfunction [19].
2.3Neuroinflammation and endothelial damageEndothelial cells are the first cells exposed to insults, such as ammonium and proinflammatory cytokines. It has been described that these cells exposed to ammonia trigger the activation of NFkB, increasing the production of oxygen free radicals and nitric oxide [22]. Endotoxemia leads to an increase in the production of nitric oxide, which is associated with endothelial dysfunction, altering brain circulation. Ammonium causes an osmotic change by damaging the permeability of the blood–brain barrier. Therefore, it disrupts the cerebral flow through an arachidonic acid-dependent pathway [20]. These mechanisms activate the TLR4 signal pathway, causing the release of proinflammatory factors, which contribute to astrocytic edema [21].
Proinflammatory cytokines that involve liver failure do not normally pass through the blood-brain barrier. However, the increase in ammonium concentrations disrupts tight junctions between endothelial cells, opening the blood–brain barrier to the passage of those cytokines. Consequently, they activate an inflammatory endothelial response through different pathways, initiating brain production of proinflammatory cytokines which include TNF-α, IL-6, and IL-1 [22]. It has been demonstrated a synergy between hyperammonemia and inflammation. The brain's inflammatory response is associated with an increase in oxidative stress, causing a reaction known as protein nitrosation, which perpetuates this brain inflammatory cycle [23].
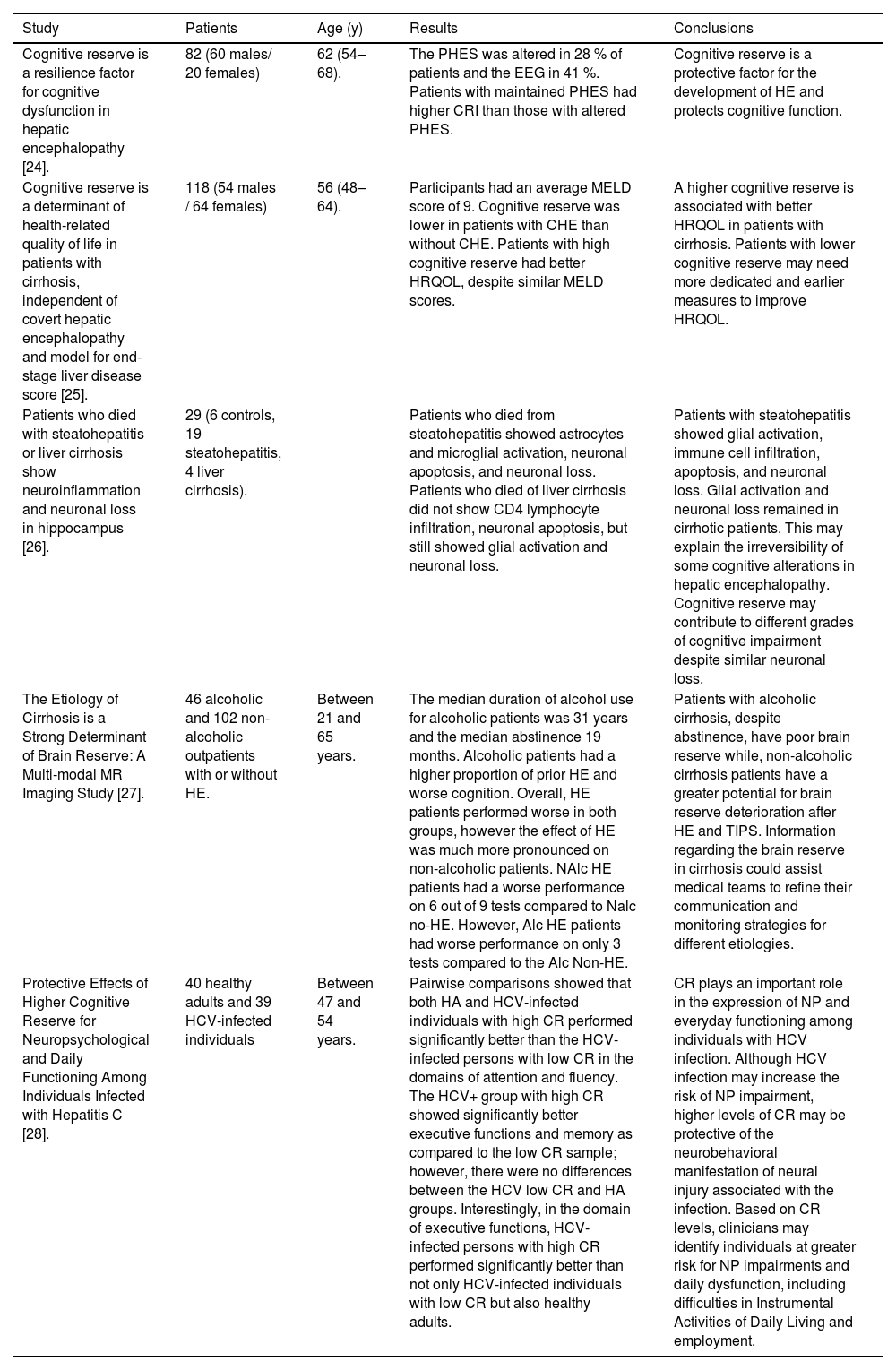
Several studies have demonstrated that cognitive reserve is a protective factor against the development of hepatic encephalopathy. It is associated with a better quality of life and its deterioration can occur in other liver-related diseases or conditions. A summary is provided in Table 1.
Relationship of cognitive reserve in multiple types of studies.
| Study | Patients | Age (y) | Results | Conclusions |
|---|---|---|---|---|
| Cognitive reserve is a resilience factor for cognitive dysfunction in hepatic encephalopathy [24]. | 82 (60 males/ 20 females) | 62 (54–68). | The PHES was altered in 28 % of patients and the EEG in 41 %. Patients with maintained PHES had higher CRI than those with altered PHES. | Cognitive reserve is a protective factor for the development of HE and protects cognitive function. |
| Cognitive reserve is a determinant of health-related quality of life in patients with cirrhosis, independent of covert hepatic encephalopathy and model for end-stage liver disease score [25]. | 118 (54 males / 64 females) | 56 (48–64). | Participants had an average MELD score of 9. Cognitive reserve was lower in patients with CHE than without CHE. Patients with high cognitive reserve had better HRQOL, despite similar MELD scores. | A higher cognitive reserve is associated with better HRQOL in patients with cirrhosis. Patients with lower cognitive reserve may need more dedicated and earlier measures to improve HRQOL. |
| Patients who died with steatohepatitis or liver cirrhosis show neuroinflammation and neuronal loss in hippocampus [26]. | 29 (6 controls, 19 steatohepatitis, 4 liver cirrhosis). | Patients who died from steatohepatitis showed astrocytes and microglial activation, neuronal apoptosis, and neuronal loss. Patients who died of liver cirrhosis did not show CD4 lymphocyte infiltration, neuronal apoptosis, but still showed glial activation and neuronal loss. | Patients with steatohepatitis showed glial activation, immune cell infiltration, apoptosis, and neuronal loss. Glial activation and neuronal loss remained in cirrhotic patients. This may explain the irreversibility of some cognitive alterations in hepatic encephalopathy. Cognitive reserve may contribute to different grades of cognitive impairment despite similar neuronal loss. | |
| The Etiology of Cirrhosis is a Strong Determinant of Brain Reserve: A Multi-modal MR Imaging Study [27]. | 46 alcoholic and 102 non-alcoholic outpatients with or without HE. | Between 21 and 65 years. | The median duration of alcohol use for alcoholic patients was 31 years and the median abstinence 19 months. Alcoholic patients had a higher proportion of prior HE and worse cognition. Overall, HE patients performed worse in both groups, however the effect of HE was much more pronounced on non-alcoholic patients. NAlc HE patients had a worse performance on 6 out of 9 tests compared to Nalc no-HE. However, Alc HE patients had worse performance on only 3 tests compared to the Alc Non-HE. | Patients with alcoholic cirrhosis, despite abstinence, have poor brain reserve while, non-alcoholic cirrhosis patients have a greater potential for brain reserve deterioration after HE and TIPS. Information regarding the brain reserve in cirrhosis could assist medical teams to refine their communication and monitoring strategies for different etiologies. |
| Protective Effects of Higher Cognitive Reserve for Neuropsychological and Daily Functioning Among Individuals Infected with Hepatitis C [28]. | 40 healthy adults and 39 HCV-infected individuals | Between 47 and 54 years. | Pairwise comparisons showed that both HA and HCV-infected individuals with high CR performed significantly better than the HCV-infected persons with low CR in the domains of attention and fluency. The HCV+ group with high CR showed significantly better executive functions and memory as compared to the low CR sample; however, there were no differences between the HCV low CR and HA groups. Interestingly, in the domain of executive functions, HCV-infected persons with high CR performed significantly better than not only HCV-infected individuals with low CR but also healthy adults. | CR plays an important role in the expression of NP and everyday functioning among individuals with HCV infection. Although HCV infection may increase the risk of NP impairment, higher levels of CR may be protective of the neurobehavioral manifestation of neural injury associated with the infection. Based on CR levels, clinicians may identify individuals at greater risk for NP impairments and daily dysfunction, including difficulties in Instrumental Activities of Daily Living and employment. |
PHES, Psychometric hepatic encephalopathy; CRI, Cognitive reserve index; EEG, Electroencephalogram; HRQOL, Health-related quality of life; Nalc, Non-alcoholics; Alc, Alcoholics; CHE, Covert hepatic encephalopathy; HCV, Hepatitis C virus; HA, Healthy adults; LT, Liver transplant; CR, Cognitive reserve; TIPS, Transjugular intrahepatic portosystemic shunt; NP, Neuroprotection; HE, Hepatic encephalopathy.
Cognitive dysfunction is linked to HE, negatively impacting health-related quality of life (HRQOL) in cirrhosis patients. Some studies suggest that poor HRQOL is associated with worse outcomes, including increased mortality [29]. HE can lead to difficulties with memory, attention, concentration, and executive functions. These cognitive deficits can hinder everyday tasks, such as managing finances, remembering appointments, and making decisions [29]. Additionally, delays in reaction times and decreased coordination can affect the ability to perform tasks requiring fine motor skills. This condition can also cause sleep disorders, including insomnia and altered sleep-wake patterns, further contributing to fatigue, and reducing daytime functioning. Impairments in cognitive and motor functions can make it challenging for patients to live independently, often necessitating assistance with daily living activities [30]. Consequently, a higher cognitive reserve is linked to better HRQOL in cirrhosis patients, regardless of disease severity and prevalence. Individuals with strong cognitive reserve can better manage the challenges of HE patients, resulting in an improved quality of life [25].
3.2Connections between different liver conditionsLiver damage, particularly in conditions like hepatitis, cirrhosis, or liver failure can have a significant implication for cognitive function and brain health, which involves several mechanisms. Hepatitis C virus (HCV) may impact cognitive reserve, including neuroinflammation, neurotoxicity, and neuroinvasion. Chronic HCV has been associated with cognitive impairment and alterations in brain structure and function, which can influence cognitive reserve [31]. Advanced stages of metabolic-associated steatotic liver disease (MASLD) are characterized by liver inflammation, fibrosis, and compromised liver function, particularly in the setting of diabetes mellitus [32]. These alterations lead to the accumulation of toxins in the bloodstream, which can exert neurotoxic effects on the brain and contribute to cognitive impairment [33]. Liver failure, whether acute or chronic, can have profound effects on cognitive function and brain reserve. The liver's ability to metabolize toxins is impaired, resulting in the accumulation of substances such as ammonia in the bloodstream. Elevated ammonia levels can have neurotoxic effects, contributing to cognitive impairment and HE. These factors affect cognitive function and quality of life, influencing brain reserve [5,6].
Patients with cirrhosis commonly face cognitive impairment before undergoing liver transplant (LT) [34]. However, LT does not always restore normal cognitive function, and when neurological recovery does happen, the duration and extent of improvement can vary significantly [35]. Cognitive impairment after liver transplant can be influenced by postoperative complications, the stress of recovering from a major surgery, and introduction of powerful immunosuppressants and antimicrobials [36,37].
4Insights from cognitive, neuroimaging, and genetic assessmentsEvaluating brain reserve involves the structural and functional aspects of the brain that support its resilience against damage and uphold its functionality. This assessment includes a combination of cognitive evaluations, neuroimaging methods, and consideration of lifestyle and biological factors. While neuropsychological tests are more directly related to cognitive reserve, they can also provide indirect information about brain reserve by evaluating the brain's ability to perform various cognitive functions [38]. Magnetic resonance imaging scans provide detailed images of brain anatomy, allowing for the measurement of brain volume, cortical thickness, and hippocampal size. These parameters serve as indicators of the brain's structural integrity. Additionally, functional MRI tracks brain activity by detecting alterations linked to blood flow, enabling the evaluation of functional connectivity and brain activation patterns during cognitive tasks [39]. Certain genetic factors, such as the APOE ε4 allele, have the potential to impact brain reserve. Understanding an individual's genetic predisposition can provide insights into their inherent brain resilience. Blood tests and cerebrospinal fluid analysis for biomarkers such as amyloid-beta, tau proteins, and inflammatory markers can provide information about brain health and potential reserve [40].
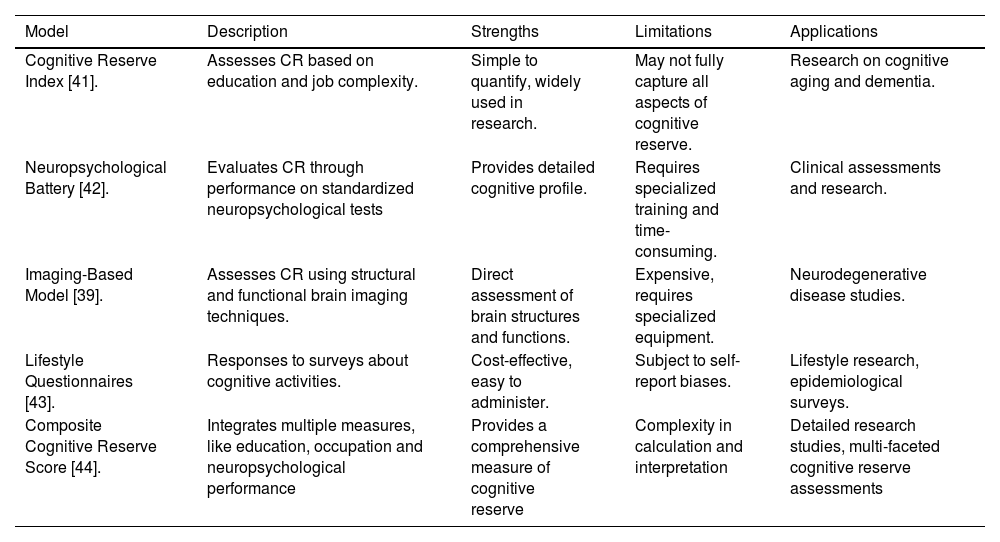
Different models have been implicated to evaluate and quantify the importance of cognitive aging and resilience. Cognitive Reserve Index (CRI), which assesses a measure of educational and occupational attainment to estimate cognitive reserve, and neuropsychological batteries that evaluates cognitive performance across different domains. To demonstrate a comprehensive overview of these different models, a table will be presented that summarizes strengths, limitations and applications (Table 2).
This table aims to elucidate the methodologies used to assess cognitive reserve, facilitating a clearer understanding of how each model contributes to our knowledge of cognitive resilience and its implications for neurodegenerative diseases.
| Model | Description | Strengths | Limitations | Applications |
|---|---|---|---|---|
| Cognitive Reserve Index [41]. | Assesses CR based on education and job complexity. | Simple to quantify, widely used in research. | May not fully capture all aspects of cognitive reserve. | Research on cognitive aging and dementia. |
| Neuropsychological Battery [42]. | Evaluates CR through performance on standardized neuropsychological tests | Provides detailed cognitive profile. | Requires specialized training and time-consuming. | Clinical assessments and research. |
| Imaging-Based Model [39]. | Assesses CR using structural and functional brain imaging techniques. | Direct assessment of brain structures and functions. | Expensive, requires specialized equipment. | Neurodegenerative disease studies. |
| Lifestyle Questionnaires [43]. | Responses to surveys about cognitive activities. | Cost-effective, easy to administer. | Subject to self-report biases. | Lifestyle research, epidemiological surveys. |
| Composite Cognitive Reserve Score [44]. | Integrates multiple measures, like education, occupation and neuropsychological performance | Provides a comprehensive measure of cognitive reserve | Complexity in calculation and interpretation | Detailed research studies, multi-faceted cognitive reserve assessments |
Hepatic encephalopathy (HE) is closely associated with delayed memory and a reduced executive function. Dementia could be masked in cirrhotic patients due to underlaying hepatic encephalopathy [45]. However, similar transcriptome profile with proteins coding RNAs involved in neuroprotection and neuro differentiation were found to be similar in Alzheimer disease and HE in animal model, implicating that AD and HE might share similar molecular mechanisms [46]. This could also be related to the brain gut axis strengthening the idea that gut microbiota has a fundamental role in brain health [47].
6Strategies through lifestyle modificationsIt is recommended to regularly engage in a variety of mentally stimulating tasks, such as reading, writing or arts. Doing these activities daily or several times a week, helps to continuously stimulate the brain. This consistent stimulation is important because it reinforces neural connections and promotes neuroplasticity. The brain's ability to adapt and reorganize itself enhances brain reserve through cognitive activities, especially with frequent participation. Individuals who consistently engage in leisure activities exhibit better cognitive outcomes compared to those who participate sporadically, which does not provide the same level of continuous activation. This irregular pattern may not be sufficient to produce lasting benefits for cognitive health. Research suggests that the key to enhancing cognitive reserve lies in the novelty and variety of activities, including reading, using computer, board or card games, mahjong, participating in forums or discussions, writing, calligraphy and painting, handicraft, playing musical instruments, investment in stock market, and gambling [48]. Engaging in tasks that challenge your brain in new ways can significantly boost cognitive function [49]. Also, the neuronal response can be improved by social activities, including community engagement and spending time with friends and family. These activities are beneficial for cognitive reserve and help to reduce the risk of cognitive decline by providing emotional support and mental stimulation. The preventive effect is attributed to the enhancement of cognitive reserve, which acts as a buffer against neurodegenerative diseases [50].
6.1Physical activityBrain health is optimized and protected by performing aerobic physical activity. It increases neuroplasticity by improving brain perfusion, reducing oxidative stress, decreasing inflammatory response, stimulating the secretion of neurotrophic brain cell factors, and diminishing exposure to neurotoxicity. Additionally, it has been demonstrated that brain health slowly reduces the loss of gray matter and hippocampal atrophy related to advanced age and cerebral insults, preserving neurons and synapses, and increasing prefrontal gray matter volume [51]. It has been shown that using smart cell phone applications in neurorehabilitation has improved executive cognitive processes and recovers the performance of complex tasks [52].
6.2NutritionValine, leucine, and isoleucine are amino acids (BCAA), substrates of different metabolic processes. Their intake contributes to the purification of ammonia in skeletal muscle. It is metabolized to glutamate incorporating ammonium into the amidation process. Isoleucine is metabolized to acetyl-CoA and succinyl-CoA which produces ATP stimulating the TCA. Another important function of BCAA is the reduction of aromatic amino acids reuptake at the brain level, which decreases the production of false neurotransmitters, such as octopamine and phenylethylamine, stimulating liver regeneration, and improving cerebral perfusion [53].
The administration of probiotics is associated with an increase in gut bifidobacteria and a decrease in ammonium concentrations and intestinal permeability. They improve the integrity of the intestinal epithelium and enhance the liverʼs ability to detoxify ammonium by decreasing endotoxemia, inflammation, and production of bacterial toxins [54].
The administration of probiotics, omega fatty acids, and zinc supplementation play distinct interrelated roles in promoting gut health, cognitive function, and overall neurological well-being.
6.3Omega fatty acidsThey represent a protection factor of cognitive reserve, decreasing the speed of cognitive loss and preventing neurodegenerative diseases in genetically predisposed patients. Fish product supplementation, rich in polyunsaturated acids, reduces cognitive alterations, improving the brain reserve [55]. Omega 5 is a neuroprotector that minimizes the cerebral injury of lipid peroxidation and oxidative stress, diminishing the activity of acetylcholinesterase [56]. The grenade polyphenols could help in anxiety-depressive disorders and memory loss. They have antioxidants and anti-inflammatory effects, that improve cerebral activity, decreasing the production of amyloid oligomers and the deposit in the CNS [57]. On the other hand, the intake of trans-fatty acids and saturated fats increases the risk of presenting cognitive alterations and dementia [58,59].
6.4ZincIt has important roles in cognitive activity, such as the activation of neuronal survival pathways, sensory processing, and neurotransmission [60]. Low levels of zinc could affect hepatocyte functions and immune response in patients with chronic liver disease [61]. Supplementation of zinc prevents cognitive alterations and neurological deficits. It reduces levels of ammonium in neuropsychological tests and produces the improvement of hepatic function, with a low dose ingestion of zinc sulfate (50 mg) daily [62]. It is an enzymatic cofactor in the urea cycle, and its deficiency causes a decrease in the activity of glutamine synthetase enzyme, which is responsible for the ammonium detoxification cycle, leading to a beneficial effect in hepatic encephalopathy patients [63].
7Therapeutic management and prevention7.1RifaximinThis broad-spectrum antibiotic represents the first-line treatment for hepatic encephalopathy. It mainly functions at gastrointestinal levels with low systemic absorption, decreasing side effects. [64,65]. A preclinical study investigated the combined effect of rifaximin and probiotics on the neurometabolic profile and glutamate levels in a rat model of type C HE. The study found that while rifaximin alone did not significantly alter the neurometabolic profile, the combination with the probiotic Vivomixx attenuated the neurometabolic alterations typically observed in bile duct ligated rats. Specifically, the increase in glutamine was less pronounced, and the reductions in glutamate and creatine levels were milder. These findings suggest potential clinical relevance for using both rifaximin and probiotics in treating HE [71]. There is a study that reviewed the combined effects of probiotics and rifaximin on gut microbiota modulation, a key factor in HE. It emphasized that probiotics and rifaximin could work synergistically to restore a healthier gut microbiota composition, which is crucial for managing HE. This study suggested that these interventions might help in mitigating the neuropsychiatric symptoms linked to HE, although it did not thoroughly address specific neurometabolic profiles [66].
7.2AntioxidantsThe use of vitamins exerts a protective effect against damage to the liver and brain, reducing oxidative stress [67]. The benefits of silymarin and l-methionine on the modulation of brain neurotransmitters have previously been demonstrated. They protect against memory impairment, decrease serotonin reuptake, help to preserve the integrity of cortical brain neurons, and reduce inflammation of microglia due to an antioxidant effect [68].
A study about animals was conducted to evaluate the effect of N-acetylcysteine (NAC) in the treatment of patients with hepatic encephalopathy. It was observed that NAC restored mitochondrial function in cases of acetaminophen-induced liver failure by replenishing mitochondrial ATP levels, reestablishing the energetic state, and exerting a neuroprotective effect [69]. Silymarin is well-known for its hepatoprotective properties. It causes antioxidant and anti-inflammatory effects, modulates liver biochemical markers, and enhances the expression of protective factors such as Nrf2. It has shown efficacy in reducing liver damage in various animal models [70]. Melatonin, a neurohormone involved in circadian rhythm synchronization and a potent immunomodulator, may act as a protector of brain reserve and reduce neurodegenerative damage. It has been described that melatonin inhibits NMDA receptors and mediates dopamine/cAMP signaling modulating dopaminergic neurotransmission, as well as the cholinergic/serotonergic system promoting GABA neurotransmission, these effects could exert a neuroprotective action increasing brain reserve [71].
7.3BezafibrateIt is a PPAR agonist that improves mitochondrial function, decreases the loss of brain reserve, and increases oxidative phosphorylation capacity, which mitigates neuroinflammation and improves cognitive functions [72]. Preclinical studies have shown that PPAR agonists can significantly improve mitochondrial function, reduce neuroinflammation, and enhance cognitive functions. PPAR-α and PPAR-γ agonists have demonstrated neuroprotective effects in models of neurodegenerative diseases like Alzheimerʼs and Parkinsonʼs di. These agonists enhance oxidative phosphorylation capacity and decrease the loss of brain reserve, which collectively contribute to mitigating neuroinflammation and improving cognitive functions [73]. Research on bezafibrate, a pan-PPAR agonist, indicates potential benefits for mitochondrial function, neuroinflammation, and cognitive functions in humans. Bezafibrate activates all three PPAR subtypes (α, β/δ, and γ), which are crucial for mitochondrial biogenesis and function. This has been shown to improve mitochondrial activity and oxidative phosphorylation, potentially offering neuroprotection and cognitive benefits [74].
7.4Noncompetitive n-methyl-D-aspartate glutaminergic (NMDA) receptors antagonistsChronic hyperammonemia lead to the activation of NMDA receptor causing a reduction in neuronal nitric oxide (NO) synthase and the impairment of glutamate NO Guanosine 3’,5’-cyclic monophosphate pathway. NMDAR are a subfamily of glutamate receptors that display physiological activities in cortical neurons increasing Ca2+ permeability. Few studies have been published on the effect of NMDAR channel blockers in hepatic encephalopathy. Memantine, broadly use for Alzheimer a neurodegenerative disease, has shown to enhance desensitization of GluN1/2A receptors of cortical pyramidal neurons reducing Ca2+ accumulation, it has been hypothesized that this could confer a neurological protection [75]. It has been reported that memantine could decrease ammonia concentration improving hepatic encephalopathy by reducing excessive glutamate production with consequent reduction of neurotoxicity [76,77]. Blocking NMDAR with memantine have been demonstrated in animal models to increase survival by reducing brain herniation due to intracranial hypertension and preventing ammonia induced neuronal death [78–80]. These effects could be dual in the brain, but also in the kidney increasing GFR leading to ammonia elimination [81], likewise protecting kidney function by reducing lipopolysaccharide induced tubular damage [82].
7.5Dopamine agonists agentsDopamine agonist agents such as levodopa/carbidopa and bromocriptine, widely used for Parkinson disease, have been anciently studied for the treatment of hepatic encephalopathy. Few clinical trials have been performed to evaluate their effect in this entity, hence, a meta-analysis showed no evidence to recommend or not their use [83].
8ConclusionsThe brain reserve acts as a protective factor against cognitive decay, reflecting the ability to optimize or maximize performance through differential recruitment of brain networks, which reflect the use of alternate cognitive strategies. Lifelong cognitive stimulation, which determines cognitive reserve, is a resilience factor for cognitive manifestations of hepatic encephalopathy. Vice versa, low cognitive reserve is a risk factor for the cognitive manifestations of covert hepatic encephalopathy, so it is essential to promote mechanisms that stimulate brain reserve prevention and slow down neuronal deterioration by improving cerebral function.
This research did not receive any specific grant from public, commercial, or not-for-profit funding agencies.



