




Non-alcoholic fatty liver disease (NAFLD), recently redefined as metabolic dysfunction-associated steatotic liver disease (MASLD), represents a significant and growing global health concern, affecting approximately 30 % of the population. The prevalence and progression of MASLD—from asymptomatic fatty liver to non-alcoholic steatohepatitis (NASH), cirrhosis, and even hepatocellular carcinoma (HCC)—underscore the urgent need to understand its underlying mechanisms [1].
A key theory, the "multiple hits" hypothesis, captures the complex etiology of MASLD, with oxidative stress as a central factor in liver injury and disease progression. This stress stems from an imbalance between reactive oxygen species (ROS) and antioxidant defenses, leading to cellular damage and worsening disease severity [2]. Our understanding has shifted to recognize MASLD as a multi-systemic disorder rooted in metabolic dysfunctions like obesity, insulin resistance, and metabolic syndrome, as highlighted in its reclassification at the 2023 EASL conference [3].
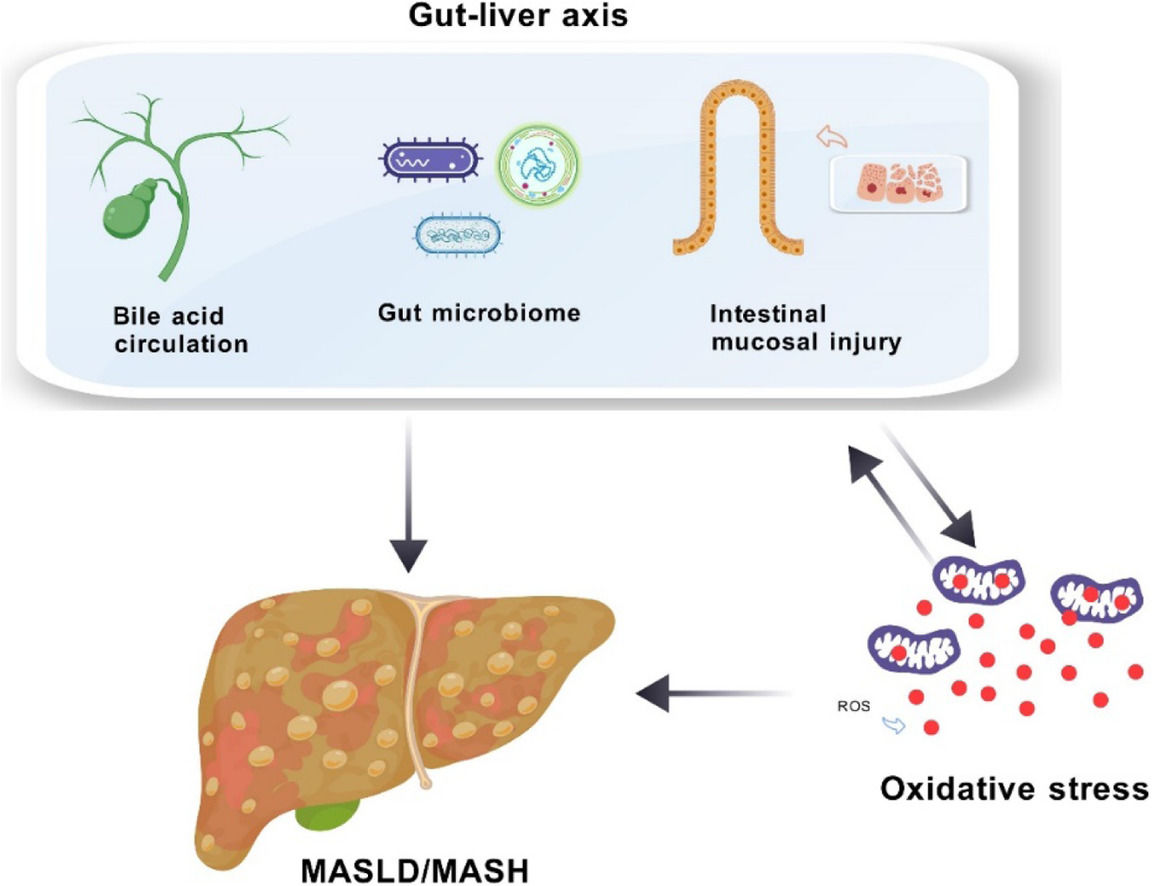
Central to MASLD research is the concept of the "gut-liver axis," which emphasizes the intricate interactions between the liver and the gut. This axis comprises cellular and molecular pathways that ensure these organs function in harmony. Bile acids (BAs), for instance, play a critical role by influencing ROS production in Kupffer cells and inducing lipid peroxidation, ultimately contributing to oxidative stress. High levels of BAs can also disrupt gut balance, potentially allowing harmful substances to cross into the liver and further escalate oxidative damage through gut microbiota alterations and increased intestinal permeability [4,5].
In this review, we provide an in-depth analysis of oxidative stress and the gut-liver axis in MASLD, examining the signaling mechanisms underlying these interactions. We also discuss the implications for disease pathogenesis and explore therapeutic strategies targeting these pathways, offering potential new approaches for MASLD management.
2Pathological Mechanisms of metabolic liver diseaseThe hallmark of MASLD is the significant lipid accumulation due to insulin resistance, excluding secondary causes like viral hepatitis and alcohol abuse. MASLD is intricately connected to metabolic syndrome, a cluster of conditions including insulin resistance, obesity, hypertension, and dyslipidemia. Type 2 diabetes emerges as a critical risk factor for the advancement to severe metabolic dysfunction-associated steatohepatitis (MASH), cirrhosis, and mortality, with poor glycemic control further exacerbating the likelihood of MASH and advanced fibrosis [6].
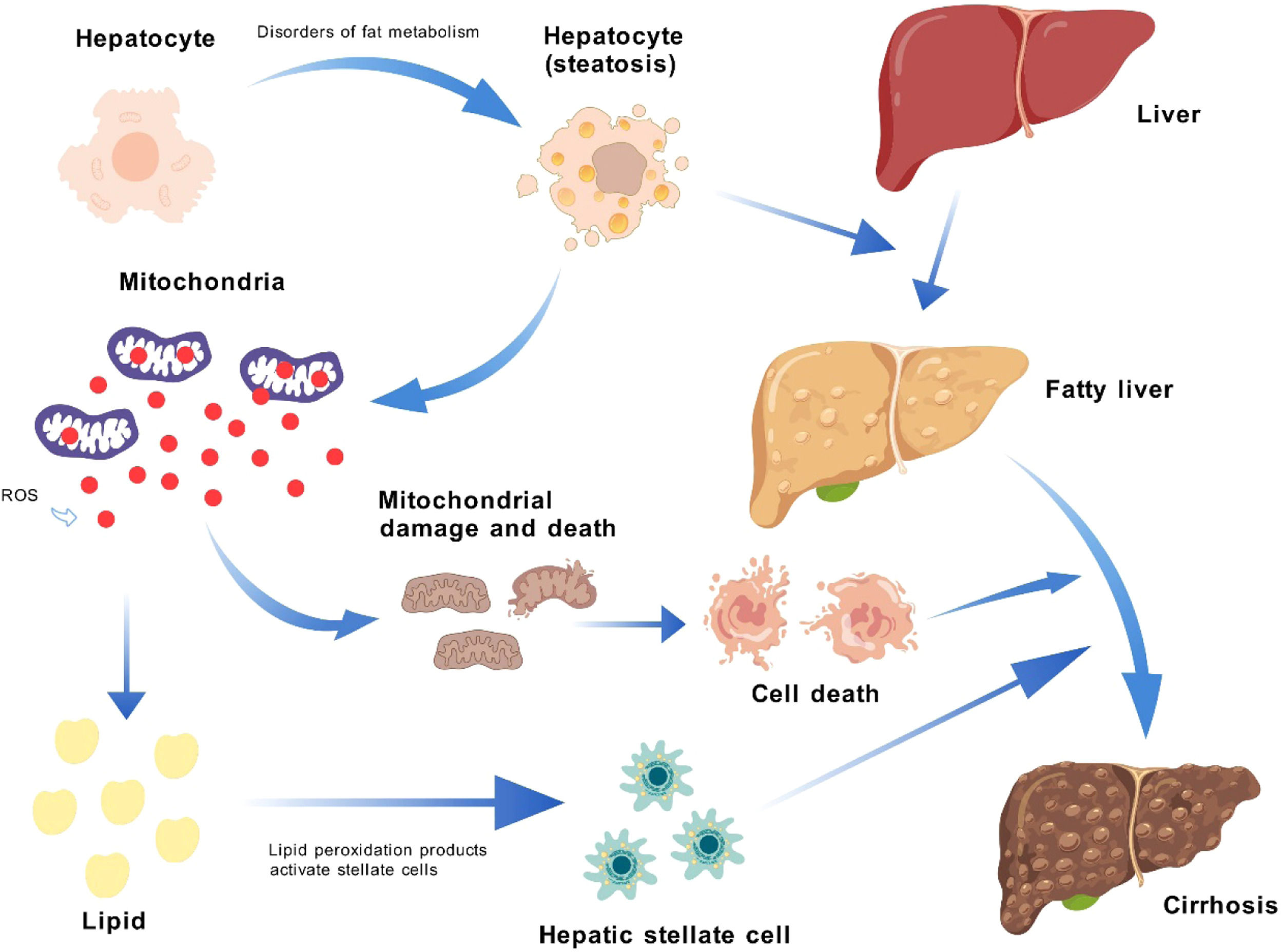
In MASLD and MASH, disruptions in lipid metabolism result in lipotoxicity and lipid peroxidation, which in turn initiate endoplasmic reticulum stress and mitochondrial dysfunction, causing liver cell damage [7]. These cellular alterations activate stem cells, which contribute to inflammation and fibrosis. The liver's response to increased energy needs involves heightened mitochondrial respiratory chain activity, leading to greater ROS production and the engagement of antioxidant defenses [8]. An overabundance of free fatty acids (FFAs) exacerbates lipid accumulation, promoting apoptosis and disrupting mitochondrial autophagy via c-Jun N-terminal kinase -dependent pathways [9]. As MASLD advances to MASH, inflammation and oxidative stress create a detrimental cycle that can damage cellular components, including the mitochondrial respiratory chain and antioxidant systems [7]. The liver's response to excessive inflammation, oxidative stress, and apoptosis includes the activation of hepatic stellate and stem cells to repair damage, a process that, while protective, can also drive liver fibrosis progression [10] (Fig.1).

Impact of Metabolic Disorders on Liver via ROS Production. Factors like insulin resistance, obesity, hypertension, and dyslipidemia disrupt lipid metabolism, causing lipid accumulation in hepatocytes and transforming a normal liver into a fatty liver. This shift increases mitochondrial respiratory activity and ROS production, which damages mitochondria and induces lipid peroxidation. The resulting ROS and peroxidation products activate hepatic stellate cells, contributing to liver fibrosis progression. ROS: reactive oxygen species.
The "multiple hits" hypothesis is a phenomenon further discovered in the study of MASLD/MASH. The "multiple hits" hypothesis regards the aggravation of fat accumulation as the first hit and oxidative stress as the second hit [11]. In current research, MASLD has been observed to exhibit a range of metabolic syndrome characteristics, from mild steatosis to systemic inflammation observed in MASH, and may even progress to liver fibrosis and HCC. In addition, inflammatory responses, changes in liver cell apoptosis signals, and the activation of stem cells may all be factors leading to the progression of mild MASLD cases to more severe disease forms, including MASH [12]. Recent research reports increasingly point to the third hit, involving the disorder of lipid metabolism, mitochondrial dysfunction accompanied by reduced fatty acid oxidation, and periodic oxidative stress responses during the course of MASLD [2]. Other theories on the "multiple hits" hypothesis of MASLD further discuss the impact of lipid metabolism, intestinal barrier dysfunction, and systemic inflammation [13].
3Gut-Liver Axis3.1BAsBAs play a critical role in digestion, but their significance in the context of MASLD extends beyond mere digestion. The hepatic synthesis of BAs is a complex process involving two pathways, with the classic pathway initiated by CYP7A1 enzyme producing cholic acid (CA) and the alternative pathway generating different BA profiles. The hepatic synthesis of BAs is a complex process involving two pathways, with the classic pathway initiated by CYP7A1 enzyme producing CA and the alternative pathway generating different BA profiles. The regulation of BA transport and synthesis is crucial, as dysregulation can lead to their accumulation, which has been linked to the cytotoxic and pro-inflammatory properties observed in MASLD [14].
The metabolism of BAs is intricately linked to liver fibrosis and inflammation in MASLD, with studies showing altered BA enzyme and transporter expression resulting in increased total bile acids (TBA) in plasma [15]. High BA concentrations can directly activate inflammatory and fibrotic cells, further complicating the disease's progression [16].
BAs also function as signaling molecules, particularly as ligands for the Farnesoid X receptor (FXR), which is central to lipid and glucose metabolism, liver regeneration, and modulation of the intestinal microbiota. The activation of FXR is instrumental in maintaining liver health and preventing fibrosis, and therapeutic interventions targeting the FXR- Fibroblast growth factor (FGF) 15/19 pathway have shown promise in MASLD treatment [17].
3.2Gut microbiotaThe gut microbiota's complex ecosystem is vital for nutrient absorption and immune system development [18]. Emerging research indicates that the gut microbiota significantly influences the development and progression of MASLD through the gut-liver axis. Changes in microbiota composition can affect BA structure and signaling, potentially contributing to disease advancement [19]. Disturbances in key gut microbiota groups have been linked to immune dysfunction and disruptions in nutrient maintenance, which may impact MASLD development [20]. Additionally, dietary factors can alter gut microbial diversity and BA metabolism, influencing the risk of MASLD [21].
3.3Intestinal mucosal immune systemThe intestinal mucosal immune system, composed of the epithelium, lamina propria, and Peyer's patches, is essential for maintaining intestinal health and preventing microbial translocation [22]. The epithelium's barrier function is crucial, and its disruption can lead to increased mucosal permeability and inflammation, a risk factor for MASLD [23,24]. The immune response involving B and T cells, as well as other immune cells, plays a significant role in MASLD's immunopathology [25,26]. The interplay between intestinal mast cells and the release of cytokines and proteases can further modulate the intestinal barrier's stability and contribute to the disease's progression [27].
4The Onset of oxidative stress4.1The production and clearance of ROSROS, encompassing a range of oxygen-free radicals and peroxides, are integral to cellular signal transduction, metabolic regulation, and inflammation [28]. Their production, primarily through the electron transport chain and other cellular processes, is a normal part of metabolism. However, an overabundance of ROS can disrupt cellular homeostasis, leading to oxidative stress and inflammation, which are hallmarks of MASLD [29]. To counteract the generation of ROS and maintain cellular homeostasis, the body has an antioxidant defense system that eliminates ROS through multiple pathways: 1) Superoxide dismutase (SOD) catalyzes the dismutation of superoxide radicals; 2) Catalase (CAT) decomposes H2O2; 3) Glutathione peroxidase (GPX) and peroxiredoxins (PRDX) degrade H2O2; 4) The thioredoxin redox cycle; 5) Glutathione S-transferase (GST) detoxifies exogenous substances. These processes work together to protect cells from oxidative damage[30].
4.2Mitochondrial oxidationMitochondria are both a source and target of ROS, with the electron transport chain being a significant site of ROS generation [31]. In MASLD, increased fatty acid oxidation can lead to excessive ROS production, particularly when the electron transport chain's capacity is not scaled up accordingly. This overproduction of ROS can impair mitochondrial function, leading to further oxidative stress and inflammation [32]. The mitochondria's permeability transition pore (mPTP) plays a critical role in the cellular response to oxidative stress, with its opening potentially initiating apoptosis and contributing to disease progression in MASLD [33] These processes are crucial in MASLD progression, highlighting the importance of maintaining mitochondrial integrity and preventing excessive ROS production.
4.3Endoplasmic reticulum stressThe endoplasmic reticulum (ER) is central to protein synthesis and is also a source of ROS, particularly under conditions of stress such as those found in MASLD [34,35]. ER stress, triggered by the accumulation of misfolded proteins due to lipid toxicity, can activate the unfolded protein response (UPR), which can lead to apoptosis and inflammation. The interplay between the ER and mitochondria through calcium signaling is crucial for cellular homeostasis and is implicated in the pathophysiology of MASLD [35].
4.4Alterations in nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX)NOXs are a family of enzymes that directly produce ROS and are closely associated with the pathophysiology of liver diseases, including MASLD. Increased NOX activity, particularly that of the NOX4 subtype, has been linked to oxidative stress, lipid peroxidation, and fibrosis in the context of liver disorders [36]. Furthermore, high-fat Western diet-fed mice exhibit increased liver NOX2 subunit expression, which correlates with hepatic steatosis severity [37]. These findings underscore NOX and its ROS as vital regulators in liver disease initiation and progression, modulating NOX activity presents a potential therapeutic strategy for managing oxidative stress in MASLD.
5The Interrelationship between the gut-liver axis and oxidative stress5.1The impact of bile acid metabolism on oxidative stressIn MASLD patients, the gut-liver axis is disrupted, affecting bile acid (BA) balance and causing cholestasis-like effects [38]. High BA concentrations can damage cell membranes and stimulate ROS production in hepatocytes and Kupffer cells, leading to hepatocellular damage through oxidation of lipids and proteins. The liver mitochondria are the primary source of BA-mediated oxidative stress, with lipid peroxidation occurring during bile duct obstruction and BA toxicity [39]. Hydrophobic BAs can impair mitochondrial respiratory chain enzymes, reducing complexes I, III, and IV activities and affecting redox and electron transport [40]. BA cycle disruption can dysregulate liver lipid metabolism, especially in the context of insulin resistance in MASLD, and visceral adipose tissue resistance can lead to FFA accumulation in the liver [41].
High FFA levels trigger ER stress responses via inositol-requiring enzyme 1 (IRE1) and protein kinase R-like endoplasmic reticulum kinase (PERK), sensing alterations in lipid membrane fatty acid ratios. Saturated FFAs, such as palmitic or stearic acids, are more cytotoxic than unsaturated FFAs like oleic acid, inducing ER stress, disrupting calcium homeostasis, and activating mitochondrial apoptotic pathways in MASH pathogenesis [16,42]. Excessive FFAs can also cause oxidative stress and ROS production independently of ER stress, associated with fatty acid oxidation catabolic activities. Normally, FFAs are oxidized to CO2 and water, reducing hepatic fat content with low ROS levels balanced by antioxidants [43]. However, in MASH progression, FFA overload enhances β-oxidation, leading to acetyl-CoA accumulation, tricarboxylic acid (TCA) cycle impairment, and mitochondrial respiration dysregulation, resulting in decreased adenosine triphosphate (ATP) synthesis and increased ROS production. The liver's antioxidant system becomes less effective, exacerbating oxidative stress [44]. Elevated ROS can activate signaling pathways, promote lipid peroxidation in hepatocytes, and cause liver cell death, generating reactive lipid species like 4&#¿;hydroxy-2-nonenal and malondialdehyde. These metabolites enhance extracellular ROS release, increase hepatic injury, activate hepatic stellate cells, and promote extracellular matrix deposition, advancing liver fibrosis [45].
5.2The influence of the gut microbiota and mucosal permeability on oxidative stressDisruption of BAs can lead to bacterial overgrowth in the distal small intestine, altering gut microbiota composition and increasing intestinal permeability [46]. Recent comprehensive reviews have provided consolidated evidence that oxidative stress serves as a cornerstone in the pathophysiology of MASLD and liver fibrosis generation, establishing a crucial link between gut dysbiosis and liver pathology. This bidirectional relationship demonstrates how gut microbiota dysbiosis can trigger oxidative stress responses, while oxidative stress can further exacerbate gut barrier dysfunction [47]. Gut microbiota can influence lipid metabolism by downregulating fasting-induced adipose factor angiopoietin-like 4 (ANGPTL4), which inhibits lipoprotein lipase (LPL). LPL is a key enzyme that regulates fatty acid release from triglyceride-rich lipoproteins in muscle, cardiac, and adipose tissues. Increased LPL activity in adipocytes leads to enhanced fatty acid uptake and triglyceride accumulation, promoting fat storage and contributing to steatosis pathogenesis [48].
Intestinal barrier function is affected by factors such as the mucus layer, antimicrobial peptides, and tight junction (TJ) proteins, which form a selectively permeable seal between adjacent epithelial cells [49]. Gut microbiota dysbiosis could potentially disrupt the arrangement of intestinal epithelial cells by relaxing TJs, triggering an adaptive immune response. TJ proteins like zonula occludens-1 (ZO-1) are crucial for maintaining epithelial cell monolayer integrity and intercellular communication. Dysregulation in gut microbiota might suppress zonula occludens-1 (ZO-1) expression, leading to increased intestinal permeability [50].
Changes in the microbiota and increased intestinal permeability may facilitate the translocation of TMAO and LPS from Gram-negative bacteria into the portal vein and bloodstream, leading to liver inflammation and damage [51]. In mice, a diet high in trimethylamine N-oxide (TMAO) increases liver lipid peroxidation, as indicated by higher malondialdehyde (MDA) and non-esterified fatty acids (NEFA) levels, and reduces antioxidant enzyme activities like total superoxide dismutase (T-SOD) and glutathione peroxidase (GSH-Px), disrupting liver lipid metabolism and causing oxidative stress damage [52]. lipopolysaccharide (LPS) from gut microbiota alterations can enhance NADPH oxidase activity, promoting oxidative stress reactions that may contribute to diseases like MASLD [53]. LPS-induced oxidative stress in the liver can activate nuclear respiratory factor 1 (NRF-1) and mitochondrial transcription factor-A expression, depleting mitochondrial glutathione and affecting mitochondrial DNA synthesis. Mitochondria-generated ROS are key in this process, with their loss leading to delayed NRF-1 expression and cell growth. ROS signaling is crucial for liver mitochondrial biogenesis related to cell proliferation and is linked to the PI3K-Akt pathway activation [54].
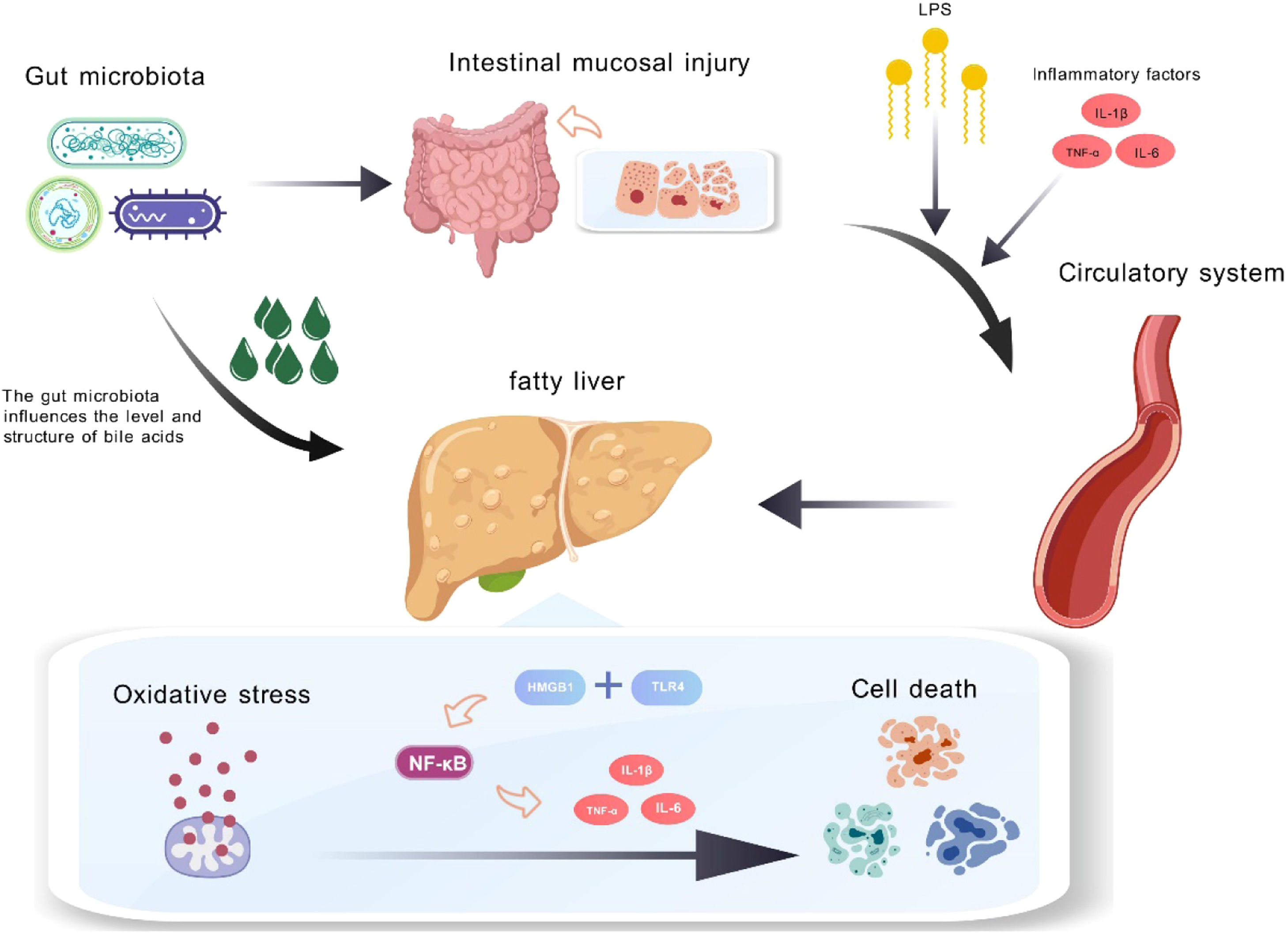
In the liver, LPS can bind to toll-like receptor 4 (TLR4) on Kupffer cells, triggering the release of inflammatory factors like tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), activating hepatic stellate cells to produce myofibroblasts that upregulate NADPH oxidase and generate reactive species, promoting oxidative stress [55]. Studies have confirmed the link between LPS and oxidative stress in various diseases. In vitro, human monocytes exposed to LPS concentrations found in atherosclerotic plaques showed increased TLR4-mediated Nox2 activity and a rise in sNox2-dp levels by about 4.5-fold [56]. This has been validated in animal models where gut microbiota alterations impact Nox2 activation and redox signaling pathways [57]. In neurodegenerative diseases, elevated Nox2 activation levels have been observed, with LPS association suggesting the gut microbiome as a significant oxidative stress source [58]. These mechanisms explain how the disrupted gut-liver axis in MASLD patients can induce oxidative stress and worsen disease onset and progression (Fig. 2).
 and inflammatory factors (TNF-α, IL-1β, IL-6) to enter the bloodstream and reach the liver. Additionally, altered gut microbiota disrupts bile acid metabolism, intensifying hepatic inflammation and oxidative stress. This ROS buildup activates nuclear factor kappa B (NF-κB) signaling via TLR4, promoting inflammation and hepatocyte death.")
Impact of Gut Microbiota Disruption on Liver Inflammation and Cell Death. In MASLD patients, changes in gut microbiota led to intestinal damage and increased permeability, allowing lipopolysaccharides (LPS) and inflammatory factors (TNF-α, IL-1β, IL-6) to enter the bloodstream and reach the liver. Additionally, altered gut microbiota disrupts bile acid metabolism, intensifying hepatic inflammation and oxidative stress. This ROS buildup activates nuclear factor kappa B (NF-κB) signaling via TLR4, promoting inflammation and hepatocyte death.
In MASLD development, increased oxidative stress intensifies lipid peroxidation, harming hepatocytes and activating stellate cells, thus accelerating liver fibrosis progression. ROS in hepatocytes also serve as signal transduction mediators, activating redox-sensitive transcription factors like nuclear factor kappa B (NF-κB), which influence hepatocyte activity regulation and inflammatory mediator production, such as TNFα and other pro-inflammatory factors [59]. ROS-damaged hepatocytes can activate the immune system via damage-associated molecular patterns (DAMPs), recruiting monocytes and neutrophils to incite inflammation. High mobility group box 1 (HMGB1), a type of DAMP, can bind to TLR4 on hepatocytes [60], activating the NF-κB pathway through a myeloid differentiation primary response 88 (MyD88)-dependent mechanism. This promotes the release of inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) and ROS, leading to intensified inflammatory responses and additional liver damage [61]. Toll-like receptor 9 (TLR9) can also bind to HMGB1, inducing similar pro-inflammatory effects by acting on dendritic cells and macrophages [62]. Cytokines like transforming growth factor beta 1 (TGF-β1) can inflict liver cell damage through death receptor signaling initiated by autocrine action and activate receptors on nearby macrophages, endothelial cells, and stellate cells through paracrine mechanisms, further promoting inflammation and fibrosis [63]. These interrelated mechanisms continually worsen MASLD's pathological state.
Recent research indicates that oxidative stress is a crucial link between intestinal abnormalities and liver pathology, playing a significant role in intestinal mucosal barrier damage mechanisms. Excessive ROS can lead to oxidative stress that disrupts cellular redox balance, damaging cellular proteins, including cytoskeletal proteins in the intestine, and ultimately leading to the destruction of the gastrointestinal barrier and increased intestinal permeability [64]. The redox-sensitive transcription factor NF-κB, present in intestinal epithelial cells, is a regulatory factor in intestinal inflammation and oxidative stress responses. The intestinal mucosa, composed mainly of intestinal epithelial cells, has NF-κB that is closely associated with gut-associated lymphoid tissue. NF-κB in the epithelium is key to maintaining the functionality and structural integrity of the intestinal epithelial barrier, enhancing antimicrobial actions and reducing oxidative stress-related gene expression [65]. An animal experiment showed that lipid peroxidation products induced the activation and upregulation of NF-κB in the intestinal mucosa of mice. While NF-κB maintains the integrity of the intestinal epithelial barrier, its overexpression can disrupt intestinal homeostasis, leading to intestinal diseases [66,67]. These mechanisms can potentially damage the intestinal barrier, allowing endotoxins and pro-inflammatory factors to enter the bloodstream, reach the liver through the portal vein system, increase oxidative stress and inflammation, cause mitochondrial dysfunction, and feedback to affect intestinal barrier function [68].
The liver and intestine are highly sensitive to oxidative stress, which has led to the development of specialized defense mechanisms to counteract excess ROS. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key player in this defense mechanism [69]. Nrf2, like NF-κB, is a redox-sensitive factor that monitors intracellular redox status and is typically bound to Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm. Upon an increase in ROS levels, Nrf2 detaches from Keap1 and moves to the nucleus, where it enhances the transcription of protective genes. By activating the antioxidant response element (ARE) in the promoter regions of downstream genes, Nrf2 triggers the expression of multiple antioxidant genes as a protective measure. This results in the transcription of various antioxidant enzymes, such as NADPH: quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), and enzymes that preserve glutathione (GSH) homeostasis [70].
May G Akl and colleagues' research has demonstrated Nrf2′s involvement in the pathophysiology of various liver diseases, including MASLD. The absence of Nrf2 can result in impeded liver regeneration, heightened oxidative stress, and increased hepatocyte apoptosis [71]. In the aforementioned animal experiments, under lipid peroxide induction, Nrf2, like NF-κB, exhibited an overexpression trend in the small intestinal mucosa. This suggests that the intestine, when faced with oxidative stress, activates certain protective mechanisms to counteract the damage [67] (Table 1).
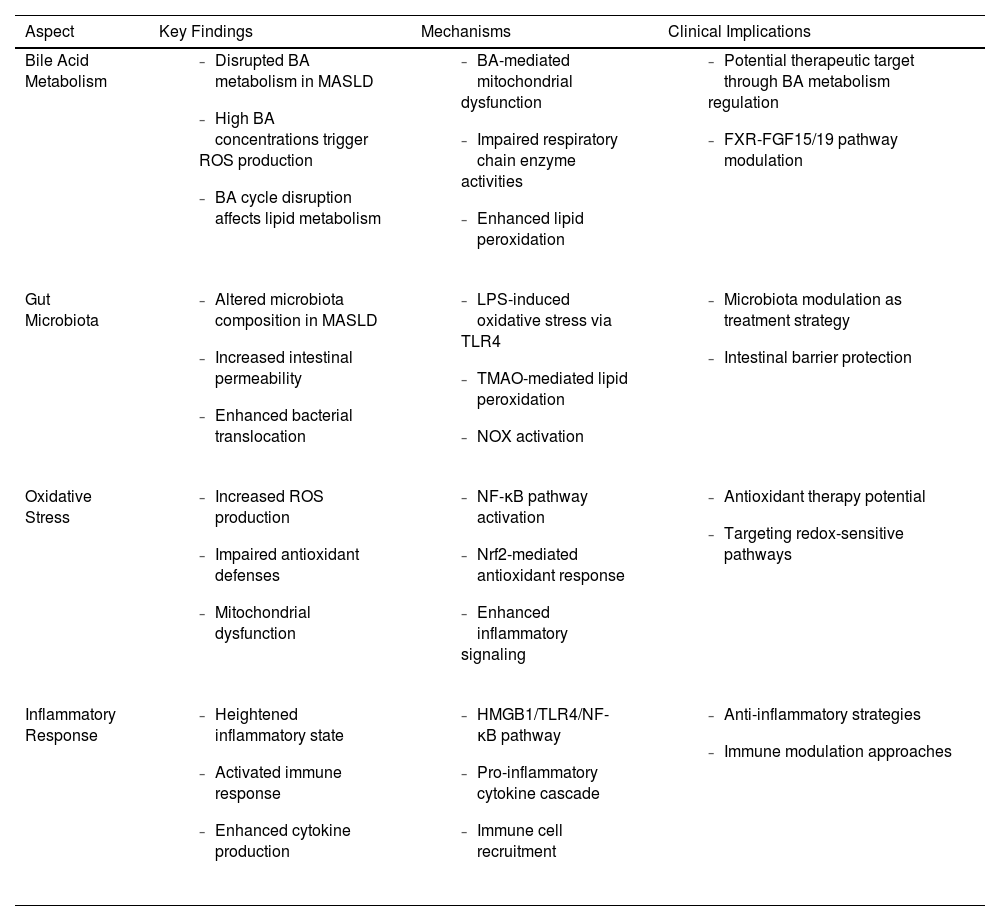
Key Mechanisms and clinical implications of gut-liver axis and oxidative stress dysregulation in MASLD.
| Aspect | Key Findings | Mechanisms | Clinical Implications |
|---|---|---|---|
| Bile Acid Metabolism |
|
|
|
| Gut Microbiota |
|
|
|
| Oxidative Stress |
|
|
|
| Inflammatory Response |
|
|
|
BA, bile acid; FGF, fibroblast growth factor; FXR, Farnesoid X receptor; HMGB1, high mobility group box 1; LPS, lipopolysaccharide; MASLD, metabolic dysfunction-associated steatotic liver disease; NF-κB, nuclear factor kappa B; NOX, NADPH oxidase; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; TLR, toll-like receptor; TMAO, trimethylamine N-oxide.
The interplay between the gut-liver axis and oxidative stress is at the core of our understanding of MASLD. This review has underscored the pivotal roles these factors play in MASLD pathogenesis, highlighting an urgent need for deeper exploration of their complex interactions.
The gut microbiome, with its significant influence on bile acid metabolism and intestinal permeability, serves as a critical element in the gut-liver axis. Disruptions to this finely balanced ecosystem can lead to adverse effects on liver health, pointing to the modulation of gut microbiota as a promising therapeutic avenue. Likewise, BAs not only facilitate digestion but also act as essential signaling molecules, influencing liver function and potentially accelerating disease progression.
Oxidative stress, resulting from an imbalance between ROS and antioxidant defenses, emerges as a primary driver of liver injury and inflammation in MASLD. The connection between ROS production and mitochondrial dysfunction, especially through redox-sensitive transcription factors like NF-κB and Nrf2, offers an intricate framework for potential therapeutic interventions in MASLD.
Moreover, oxidative stress molecules, such as MDA, HNE, and ROS, have shown strong correlations with the severity of MASLD. These biomarkers not only reflect the oxidative stress state but also align with the progression of hepatic steatosis, fibrosis, and inflammation, highlighting their diagnostic and prognostic potential. Targeting redox-sensitive pathways mediated by NF-κB and Nrf2 could provide a dual benefit—both as biomarkers for monitoring disease activity and as therapeutic targets for intervention. Exploring their application in clinical contexts may enhance precision in MASLD diagnosis and treatment.
Despite valuable insights from current research, a comprehensive understanding of these complex mechanisms remains limited. Future studies must focus on identifying and validating novel therapeutic targets, including regulators of bile acid synthesis and mitochondrial antioxidant defenses. Clinical trials aimed at addressing gut microbiota and oxidative stress in MASLD patients will be essential for translating these insights into effective clinical applications.
In conclusion, advancing MASLD research requires a multi-faceted and integrative approach—linking gut health, liver function, and oxidative stress pathways. By pursuing these interconnected avenues, we hope to develop innovative strategies that will improve MASLD treatment, ultimately benefiting patient outcomes and public health.
FundingThis research was supported by the National Natural Science Foundation of China (NO.81973598), the Chinese Medicine Clinical Research Program (NO.2024ZL103, NO.202371061), the Research Fund of National Health Commission (NO.WKJ-ZJ-2435), and the Zhejiang Provincial Traditional Chinese Medicine Science and Technology Program Young Talent Project (NO. 2021ZQ086).
Author contributionsMi Zhou and Jianyu Lv performed the selection of references, wrote and revised the manuscript and designed the figures. Data collection and analysis were performed by Xinli Chen and Yujie Shi. Guanqun Chao and Shuo Zhang provided valuable guidance and revised the manuscript. All authors have read and approved the final manuscript.
Thanks to all authors for their contributions to the manuscript.



