



La enfermedad de Gaucher (eg) es un trastorno hereditario autosómico recesivo, el cual produce alteraciones a órgano blanco como hígado, bazo, sistema nervioso, médula ósea y pulmones.
Caso clínicopaciente masculino de cinco años de edad, presenta episodios persistentes de epistaxis con dos semanas de evolución, equimosis en las extremidades y hepatoesplenomegalia, dolor en extremidades, pérdida progresiva de la fuerza muscular, regresión psicomotora desde hace 10 meses y epilepsia tipo mioclónica. Los rayos X de huesos largos asemejan forma de “Matraz de Erlenmeyer”, biopsia de médula ósea con hipercelularidad secundaria a abundantes histiocitos espumosos y datos morfológicos de acumulación lisosomal. Se integra el diagnóstico y estatificación de eg tipo iii b.
Conclusiónel tratamiento con reemplazo enzimático de imiglucerasa, topiramato, calcitriol y carbonato de calcio mejoraron la condición clínica del paciente.
Gaucher disease (gd) is an autosomal recessive hereditary disorder, which causes disturbances on target organs such as liver, spleen, nervous system, bone marrow and lungs.
Case reportA five-year-old male patient with persistent episodes of epistaxis with two weeks of progression, limb ecchymosis and hepatosplenomegaly, pain in the limbs, progressive loss of muscle strength, psychomotor regression during 10 months, and myoclonic epilepsy. The X-ray from long bones showed the shape of an Erlenmeyer flask, bone marrow biopsy with hypercellularity, due to abundant foamy histiocytes and lysosomal accumulation. We integrated the diagnosis and stratification of eg type iii b.
Conclusionenzyme replacement therapy imiglucerase, topiramate, calcitriol and calcium carbonate improved the clinical condition of the patient.
A doença de Gaucher (dg) é um desordem hereditario autossômico recessivo, que causa alteraçoes no organo branco como fígado, baço, sistema nervoso, medula óssea e pulmão.
Caso clínicom paciente do sexo masculino de cinco anos, presenta episódios persistentes de epistaxis com duas semanas de evolução, equimoses e hepatoesplenomegalia, dor nas extremidades, perda progressiva de força muscular, regressão psicomotora por 10 meses e epilepsia tipo mioclônico. Os raios-X de osssos longos se assemelhan a un “Frasco de Erlenmeyer”, biópsia da medula óssea com hipercelularidade secundária a abundantes histiócitos espumosos e dados morfológicos da acumulação lisossômica.
Conclusãoterapia con reposição enzimatica imiglucerasa, topiromato, calcitriol e carbonato de cálcio melhorou a condição clinica do paciente.
La Enfermedad de Gaucher (eg) es un trastorno hereditario autosómico recesivo;1 cuenta con una baja frecuencia en la población mundial de uno por cada 100 mil habitantes, sin embargo en los judíos Ashkenazi uno de cada 850 habitantes puede llegar a presentar dicho trastorno.2,3
La fisiopatología está basada en la alteración para la síntesis de la enzima β-glucocerebrosidasa, cuya producción es insuficiente o en ciertos casos no existe, esto impide el procesamiento adecuado del glucocerebrósido y ocasiona un almacenamiento inapropiado en los lisosomas de las células del sistema fagocítico (macrófagos y monocitos),4 con alteraciones a órganos blanco como hígado, bazo, sistema nervioso central, médula ósea y pulmones,5 que clínicamente se expresan con: hepatomegalia, esplenomegalia, desórdenes neurológicos, alteraciones del crecimiento y desarrollo neuropsicomotriz, así como trastornos de origen hemático, como coagulopatías e infecciones recurrentes secundarias a la disminución de la estirpe celular correspondiente.6
La eg se puede clasificar en tres tipos dependiendo del fenotipo y genotipo, cuya presentación clínica es específica.7
Caso clínicoPaciente masculino de cinco años de edad, procedente de una comunidad rural mexicana, que acude al servicio de urgencias por presentar epistaxis persistentes de dos semanas de evolución y equimosis en extremidades de origen no traumático. Cuenta con el antecedente de múltiples caídas, atribuidas a estrabismo del que es portador desde los dos años de edad; así como, dolor constante en extremidades y pérdida progresiva de la fuerza, sin tolerar la bipedestación por más de 15 minutos, aunado a regresión psicomotora desde hace 10 meses y epilepsia de tipo mioclónica llegando a presentar de cinco a siete episodios por día.
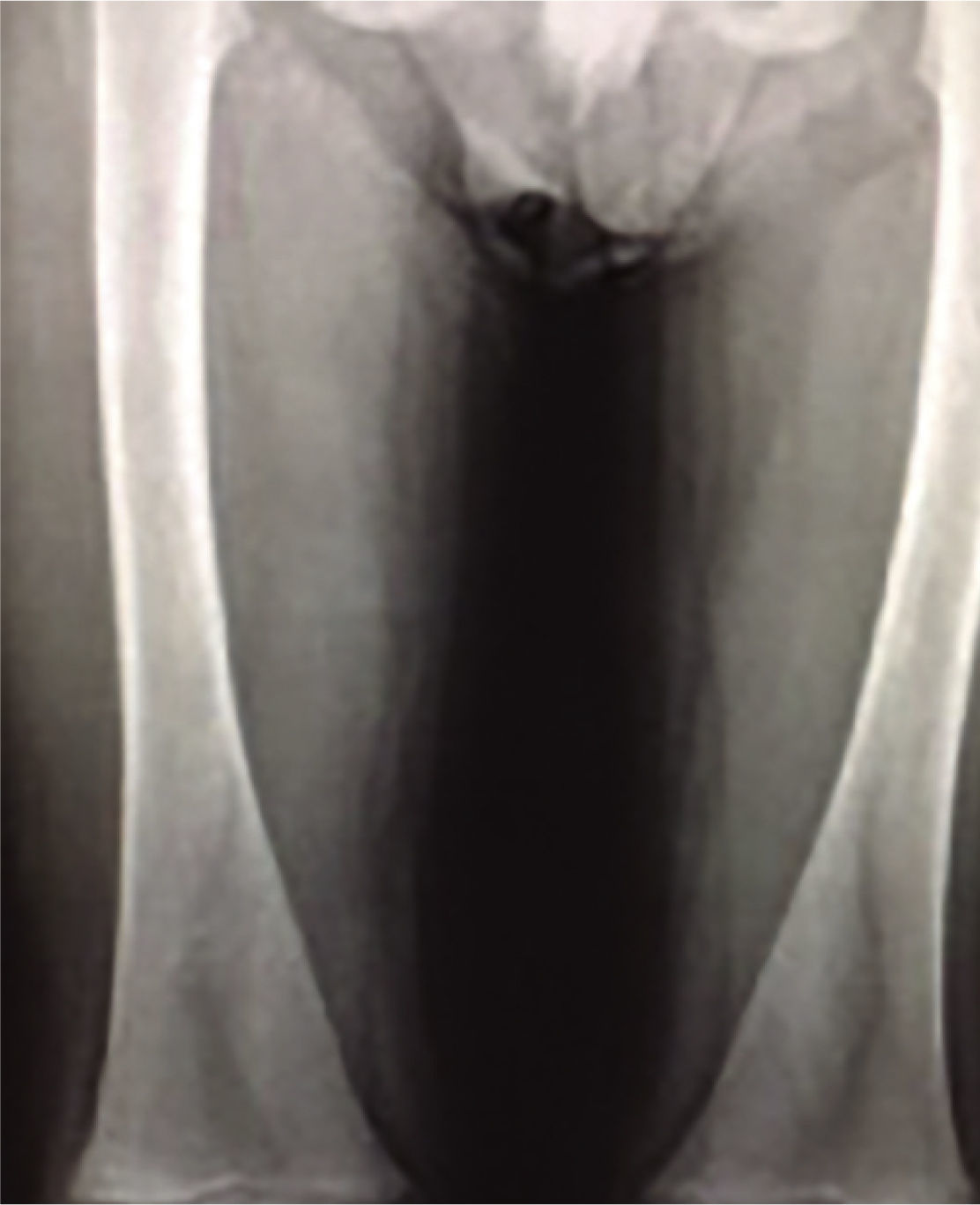
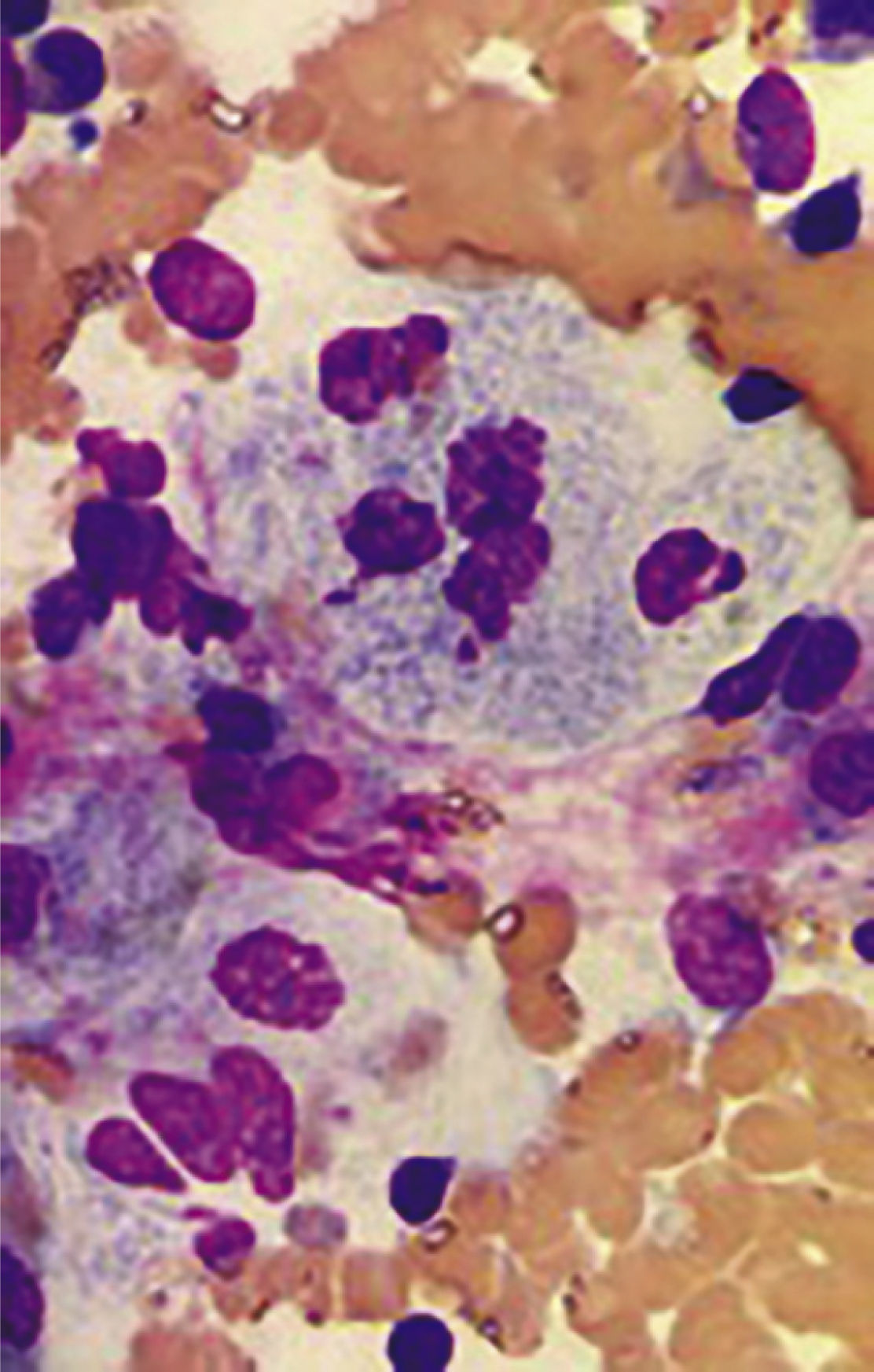
En la exploración física se encuentra un paciente alerta, inquieto, bradilálico, de edad aparente menor a la cronológica, con estrabismo divergente; es palpable el crecimiento de la glándula hepática y el bazo, rebasando el borde inferior de parrilla costal por diez y cinco centímetros respectivamente, en las cuatro extremidades son visibles múltiples equimosis con pérdida severa de la fuerza muscular. Se realizaron diversos estudios; la biometría hemática y tiempos de coagulación arrojaron los siguientes resultados: hemoglobina de 11.8 gr/dl, Hto. 35.3%, leucocitos 4.32, neutrófilos 30% del total, plaquetas 71 mil, ttp 15.2, inr 1.34, tpt 38.4. Los rayos X de proyección anteroposterior y comparativa de fémur, mostraron alteraciones en el remodelado óseo de las epífisis distales, evidenciando la deformidad característica de “Matraz de Erlenmeyer” (figura 1), la densitometría ósea reveló datos de osteopenia severa. En el aspirado y biopsia de medula ósea se evidenció aumento de la celularidad a expensas de histiocitos con citoplasma basófilo abundante, histiocitos espumosos (80%) y datos morfológicos de acumulación lisosomal (figura 2).
")

Aspirado de medula ósea con hipercelularidad a expensas de histiocitos, “células espumosas”; tinción de hematoxilina y eosina, las células violeta corresponden a elementos celulares de la médula ósea, en rosa claro corresponden a lípidos complejos en el lisosoma celular; micrografía 50X
Dada la presentación clínica se solicitaron estudios para medir la función enzimática específica asociada a enfermedad de Gaucher: determinación enzimática de β Glucosidasa Ácida en leucocitos (0.0430 nmol/mg prot/min) y actividad de β-glucocerebrosidasa (0.02μmol/L/h). En consecuencia, fue necesario realizar una prueba de biología molecular, cuyo resultado determinó la existencia de una mutación deletérea en el exón 6 (Phe213I1e) en el gen gba, hallazgo consistente para hacer el diagnóstico de enfermedad de Gaucher. A través de las manifestaciones clínicas del paciente y los estudios específicos confirmatorios se pudo estadificar una enfermedad de Gaucher tipo iii b.
Se decidió iniciar tratamiento de reemplazo enzimático con imiglucerasa y sintomático para demás padecimientos con topiramato, calcitriol y carbonato de calcio (tabla 1). En la primera evaluación, tres meses después de iniciado el tratamiento específico, se apreció mejoría gradual clínica del paciente, con regresión de la hepatoesplenomegalia y aumento de la fuerza muscular, seis meses después la biometría hemática tuvo mejoría con todos los elementos analizados encontrándose dentro de parámetros normales, regresión completa del crecimiento anormal de hígado y bazo, recuperación de la fuerza en todas las extremidades y desaparición de equimosis en piel y ausencia de epistaxis, sin embargo, las alteraciones neurológicas no presentaron mejoría.
DiscusiónLas características clínicas del caso presentado, constituyen una orientación primaria en el diagnóstico de eg. El estrabismo, hepatoesplenomegalia y deterioro neurológico son un indicador de la eg tipo iii. El hueso en forma de matraz de Erlenmeyer, es una característica que describe una alteración en el remodelado óseo del fémur, que consiste en la perdida de la concavidad de la línea metafisiaria, con un adelgazamiento pronunciado de su capa cortical, cuya presentación se asocia a trastornos como enfermedad de Gaucher y enfermedad de Niemann-Pick.8 El estudio histopatológico de medula ósea revela presencia anormal de almacenamiento lisosomal, complementado con la determinación de actividad enzimática específica, elementos necesarios que hacen posible el diagnóstico y estadificación de dicha enfermedad.9 Existe evidencia de que la suspensión abrupta del tratamiento condiciona regresión de las mejoras logradas por el medicamento, además de provocar resistencia a la terapia de sustitución enzimática en posteriores reinicios, por lo cual se sugiere continuar con el tratamiento de por vida sin interrupción, el pronóstico mejora cuando la terapia es aplicada de forma precoz en relación con el diagnóstico.10
La eg y otros desórdenes genéticos son padecimientos que en la actualidad han tenido una tendencia creciente de casos reportados en México. En ese mismo sentido, los médicos familiares tienen un papel fundamental en la identificación de enfermedades hereditarias y pueden iniciar tratamientos adecuados de manera precoz para limitar en los pacientes las secuelas de estas enfermedades con el propósito de brindarles una mejor calidad de vida.
ConclusionesEl tratamiento con reemplazo enzimático de imiglucerasa, topiramato, calcitriol y carbonato de calcio mejoraron la condición clínica del paciente. Se requiere profundizar en el entendimiento de esta patología para establecer líneas terapéuticas más efectivas para su abordaje y tratamiento.
Universidad Autónoma del Estado de México, Facultad de Medicina.
Sugerencia de citacion: Lucio-Garcia C, Noriega-Salas L. Enfermedad de Gaucher: estudio de caso. Aten Fam. 2017;24(4):176-178.