


Se desconocen las características citopatológicas de las leucemias agudas en pacientes de Chiapas, México, ya que es una población relativamente aislada con alto índice de consanguinidad, lo cual podría afectar la evolución y la respuesta terapéutica.
MétodosSe clasificaron morfológica, inmunofenotípica y genotípicamente 81 casos de leucemia aguda en pacientes atendidos en el Hospital de Especialidades Pediátricas de Chiapas, indicando riesgo al ingreso y situación al momento del estudio. Los resultados se comparan con información nacional e internacional pertinente.
ResultadosSe encontró la siguiente proporción de tipos de leucemia aguda: leucemias B, 75.3%; mieloides, 16%; de células T, 3.7%; B-M, 3.7% y de células NK, 1.2%. Las alteraciones genéticas estuvieron presentes en 40.6% de las B y en 69% de las mieloides. La alteración genética se relacionó con la evolución del paciente a corto plazo en las leucemias tipo B; no así en las mieloides. En las B, los casos con el gen MLL alterado fallecieron en menos de un mes, los casos con la translocación t(1;19)(q23;p13) han tenido buena evolución, y aquellos con la t(12;21)(p13;q22) han tenido mala evolución a medio plazo. La hiperdiploidía se presentó en el 20% de los casos B; el 83% de ellos permanecen en remisión de 1 a 12 meses desde el diagnóstico. El 69% de los casos con leucemias mieloides falleció o abandonó el tratamiento en recaída de 15 días a 37 meses del diagnóstico.
ConclusionesLa proporción de los diferentes tipos de leucemia aguda atendidas en el HEP es similar a la encontrada en otras partes del país. Su comportamiento y desenlace está relacionado con la presencia o ausencia de alteraciones genéticas específicas y no específicas.
Childhood acute leukemia cytological features are unknown in Chiapas, Mexico. Defining these features is important because this is a relatively isolated population with high consanguinity index, and these aspects could determine differences in responses to treatment and outcome.
MethodsEighty-one childhood acute leukemia cases treated at the Hospital de Especialidades Pediátricas in Chiapas were characterized by morphology, immunophenotype, genotype, initial risk assignment and status at the time of the study.
ResultsThe proportion of leukemic cell types found in this study was B cell, 75.3%; myeloid, 16%; T cell, 3.7% and NK 1.2%. In B cell leukemia, genetic alterations were present in 40.6% of cases and had a specific outcome regardless of initial risk assessment. Cases with MLL gene alteration died within a month from diagnosis. Translocations were present in 17.5% B cases; t(1;19) was present in those with a favorable outcome. The t(12;21) translocation was related to initial remission and midterm relapse and dead. Hyperdiploidy was present in 20% of B cell cases with good outcome. In 38.5%of myeloid cases were translocations and karyotypic abnormalities. Short-term outcome in this group has been poor; 69% have died or abandoned treatment in relapse from 15 days to 37 months after diagnosis.
ConclusionsRelative frequency of different types of acute leukemia in patients treated at a tertiary level pediatric hospital in Chiapas, Mexico, was similar to the one found in other parts of the country. Patients’ outcome, under a standardized treatment, differs according to the group, the subgroup and the presence and type of genetic alterations.
A partir de la creación del Seguro Popular de Salud (SPS) en el 20041, conocido entonces simplemente como Seguro Popular, las poblaciones autóctonas del estado de Chiapas se han ido incorporando progresivamente a este beneficio que les garantiza diagnósticos y tratamientos completos para un número creciente de padecimientos, entre ellos, los padecimientos neoplásicos en niños. Con el propósito de satisfacer la demanda creciente de atención médica derivada del SPS, en el año 2006 se creó el Centro Regional de Alta Especialidad de Chiapas, de tercer nivel, constituido por el Hospital de Especialidades Pediátricas (HEP) de Tuxtla Gutiérrez, para atender a la población infantil, y el Hospital Regional de Alta Especialidad en Ciudad Salud, Tapachula, para la población adulta2.
Con este esquema de servicios de salud, hoy es posible conocer con mayor precisión la problemática de salud de los grupos autóctonos, históricamente aislados.
Entre las enfermedades emergentes en la población infantil, las neoplásicas destacan por su impacto. Desde el inicio de actividades del HEP en el año 2006, estas enfermedades han sido el grupo de padecimientos por el que los pacientes son más frecuentemente hospitalizados, y de entre ellos, la leucemia aguda (LA) ha sido el diagnóstico de egreso más frecuente de acuerdo con el área de bioestadística del HEP.
El diagnóstico preciso de LA descansa actualmente en dos grandes pilares: la clínica y la caracterización citopatológica de las células sanguíneas malignas, tanto en sangre periférica como en médula ósea. Esto incluye la cuantificación y observación microscópica de sus características, el inmunofenotipo y los estudios genéticos (índice de DNA, cariotipo y translocaciones moleculares) en médula ósea. A la fecha no existe un reporte detallado de estas características en la población de pacientes con LA atendidos en el HEP.
A partir de finales del 2013 (fecha en la que se dispuso de un citómetro de flujo en el HEP) ha sido posible llevar a cabo el estudio de los casos sospechosos de leucemia en el hospital, y definir con mayor exactitud las características inmunofenotípicas de las células malignas (i.e., la línea y subpoblación celular afectada, su grado de diferenciación y la expresión de marcadores aberrantes). Por otro lado, los avances en el conocimiento y estudio del genoma humano en diversas patologías permiten la identificación de translocaciones y alteraciones cariotípicas diversas, específicas y no específicas, y su relación con los diferentes tipos de leucemia (definidos mediante inmunofenotipo). La suma de estos elementos ha llevado a la identificación de subconjuntos de leucemias dentro de una misma línea celular, que permiten su individualización citopatológica, pronóstica y terapéutica. Como resultado, es posible asignar un tratamiento específico, definido en los Protocolos Nacionales de aplicación a cada paciente3,4.
En este estudio retrospectivo transversal se presentan los resultados de la caracterización citopatológica (morfológica, inmunofenotípica y genotípica) detallada de 81 casos de LA en la infancia, atendidos en el HEP de 2013 a 2016 en población mayoritariamente autóctona del estado de Chiapas. Lo anterior con el propósito de contar con la información panorámica inicial de los tipos de LA que se presentan en la población pediátrica atendida en el HEP (grupo de estudio). Adicionalmente, se presenta la información de los diversos tipos de LA identificados con el estatus clínico de los pacientes en el momento de este análisis, y su clasificación inicial de riesgo. Los hallazgos se compararon con aquellos obtenidos en otros centros nacionales e internacionales.
2MétodosSe evaluaron 81 casos consecutivos de LA entre diciembre del 2013 y junio 2016. El inmunofenotipo fue estudiado en el laboratorio de citometría del HEP. Estos representaron aproximadamente el 20% del total de casos vistos en el HEP desde el inicio de sus operaciones. Los datos adicionales fueron obtenidos de la base de datos electrónica del laboratorio del HEP (Athenea); los datos de la evolución y situación actual del paciente, de los expedientes clínicos electrónicos del Sistema de Información para la Gerencia Hospitalaria (SIGHO) del HEP.
2.1DiagnósticoEn todos los casos, el diagnóstico de LA fue realizado por un oncólogo u hematólogo pediatra del HEP y sustentado con el cuadro clínico compatible (presencia de blastos en sangre circulante, más del 30% de blastos en el aspirado de médula ósea). De forma preliminar, se clasificó la línea celular afectada y la apariencia morfológica de los blastos en linfoblástica (L1, L2 o L3) o mieloblástica (LMA).
La cuenta leucocitaria en sangre periférica se realizó con un contador electrónico de células (Advia® 120, Siemens Healthcare Diagnostics, Eschborn, Alemania), empleando la técnica recomendada por el fabricante. Los frotis de sangre y médula ósea se tiñeron con colorante Wright (genérico) y se analizaron en forma convencional, elaborando un diagnóstico inicial de probabilidad y una clasificación provisional de la leucemia de acuerdo con las características morfológicas de los blastos.
2.2InmunofenotipoEl inmunofenotipo de 67 de los 81 casos (83%) correspondió al estudio diagnóstico primario, y el de 14 (17%) al inmunofenotipo realizado en el HEP en el paciente conocido en el momento de su recaída (secundarios). En todos los casos, el inmunofenotipo se realizó en la médula ósea, empleando un citómetro BD FACSCanto II® (Becton Dickinson and Co., New Jersey, USA) equipado con láseres rojo (633nm) y azul (488nm) y anticuerpos conjugados a un fluorocromos [isotiocianato de fluoresceína (FITC), ficoeritrina (PE), proteína peridina clorofila (PerCP-Cy5.5) o aloficocianina (APC)]. Se empleó la tinción celular directa, siguiendo el procedimiento recomendado por el fabricante. Los paneles de marcadores empleados fueron diseñados internamente, tratando de incluir los anticuerpos recomendados por el consorcio EuroFlow5. El estudio de cada caso se llevó a cabo en dos rondas: en la primera, se determinó la existencia de una población clonal neoplásica y la línea celular involucrada empleando tres tubos, cada uno con una combinación de cuatro anticuerpos; en la segunda ronda se determinó la subpoblación neoplásica y su grado de diferenciación, se descartó la coexistencia de otras líneas neoplásicas y se valoró la expresión de marcadores aberrantes. Se empleó un mínimo de siete tubos por caso, cada uno, a su vez, con cuatro anticuerpos (28 determinaciones). Los anticuerpos conjugados empleados fueron dirigidos contra los siguientes antígenos: CD1a, CD2, cCD3, CD3, CD4, CD5, CD7, CD8, CD10, CD11b, CD13, CD14, CD15, CD16, CD20, CD22, CD34, CD36, CD38, CD45, CD56, CD58, CD61, CD64, CD71, cCD79a, CD117, CD123, cMPO (mieloperoxidasa citoplasmática), nTdT (desoxinucleotidil transferasa terminal nuclear), HLA-DR (antígeno leucocitario humano-DR, del inglés human leukocyte antigen-DR), cIgM κ, cIgM λ, TCR α/β y TCR γ/δ. Típicamente, la valoración de un caso de leucemia B incluyó 19 especificidades diferentes en 28 determinaciones; uno de leucemia T incluyó 21 especificidades; uno de leucemia mieloide, 20 especificidades. En el caso de esta última, el estudio habitualmente se extendió, llegando hasta 36 especificidades en 40 determinaciones. Un marcador se consideró positivo cuando reaccionó con el 20% o más de las células estudiadas. Los resultados se analizaron con el programa BD FACS Diva v.6.1.3 (BD Biosciences, Becton Dickinson and Co., New Jersey, USA).
Con los resultados del estudio se confirmó el diagnóstico inicial (clínico y de laboratorio) y se determinó con precisión la línea celular neoplásica; la expresión de antígenos aberrantes y los casos se subclasificaron, dependiendo de su estadio de diferenciación o la subpoblación afectada, con base en los lineamientos modificados del Grupo Europeo para la Clasificación Inmunológica de las Leucemias (EGIL)6,7. Los casos de LMA asignados inicialmente a los diversos subgrupos de la Clasificación Francesa-Americana-Británica (FAB) por morfología se ratificaron o rectificaron de acuerdo con el inmunofenotipo detallado, siguiendo los lineamientos de Abdul-Hamid8. Para la clasificación de las LA de fenotipo mixto (LAFM; bilineales o bifenotípicas) se siguió el criterio del Sistema de Calificación para la Leucemia Aguda Bifenotípica del mismo EGIL6.
2.3Translocaciones y otros rearreglos génicosSe analizó la médula ósea para detectar la presencia de las 26 translocaciones más frecuentes en la LA: la inversión y la deleción de DOT1L (DOT1-like, del inglés disruptor of telomeric silencing 1-like). Se empleó la técnica de RT-PCR multiplex anidada comercial (Hemavision®-28N; DNA Diagnostic, Risskov, Dinamarca) siguiendo las instrucciones del fabricante.
2.4CariotipoEn 73 casos se realizó el estudio de cariotipo en médula ósea, obteniéndose metafases analizables en 68 de los 73 casos estudiados. El cariotipo se realizó empleando la técnica convencional de cultivo no estimulado durante 24 a 48h, seguido de la adición de colchicina, solución hipotónica y solución fijadora, preparación ulterior de laminillas, tinción para bandeo G y análisis microscópico.
2.5Índice de DNAEn todos los casos se determinó el índice de DNA en las células de médula ósea mediante citometría de flujo, empleando el kit Coulter® DNA Prep™ para la extracción y tinción del DNA y el citómetro Epics XL® para su análisis (ambos de Beckman Coulter, Brea, CA, USA).
Una vez obtenidos los resultados de los estudios genéticos, se correlacionaron los subgrupos ya identificados (EGIL) con la Clasificación Internacional de Enfermedades-Oncología, versión 3 de la Organización Mundial de la Salud (OMS) (ICD-O-3)9, y se presentan en forma conjunta.
2.6EtnicidadLa determinación de etnicidad se llevó a cabo subjetivamente, considerando el grado de marginación y tamaño de la localidad de origen, la lengua dominante de los padres y la autoevaluación de los familiares responsables del paciente.
3Resultados3.1Características generalesLa edad promedio de los pacientes fue de 7 años 8 meses (IC 95% 6.7-8.7 años). La distribución de los casos por grupos de edad mostró que 31 casos (38.3%) fueron menores de 5 años, 24 (29.6%) de 5 a 9 años, 20 (24.7%) de 10 a 14 años y 6 (7.4%) de 15 a 18 años. La relación para estos casos fue de 1.3:1 (masculino/femenino), la cual no difiere de la de 1.4:1 observada en la población pediátrica no leucémica hospitalizada en el HEP en el mismo periodo, de acuerdo con las áreas de Bioestadística y Tecnologías de la Información de este hospital.
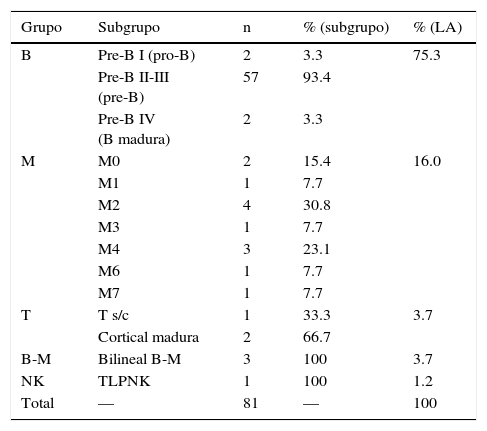
3.2Clasificación y subclasificaciónEn la tabla 1 se presentan los diagnósticos definitivos en los pacientes estudiados en orden de frecuencia con la subclasificación de acuerdo con EGIL. Puede observarse claramente que predominan las leucemias que afectan a los linfocitos B (75%), seguidas de las leucemias de línea mieloide (16%), las de fenotipo mixto (4%), las de linfocitos T (4%) y casos aislados de otras variedades. En todos los casos secundarios, el estudio coincidió con el diagnóstico primario (realizado por un proveedor externo).
Clasificación de los pacientes con leucemias agudas
| Grupo | Subgrupo | n | % (subgrupo) | % (LA) |
|---|---|---|---|---|
| B | Pre-B I (pro-B) | 2 | 3.3 | 75.3 |
| Pre-B II-III (pre-B) | 57 | 93.4 | ||
| Pre-B IV (B madura) | 2 | 3.3 | ||
| M | M0 | 2 | 15.4 | 16.0 |
| M1 | 1 | 7.7 | ||
| M2 | 4 | 30.8 | ||
| M3 | 1 | 7.7 | ||
| M4 | 3 | 23.1 | ||
| M6 | 1 | 7.7 | ||
| M7 | 1 | 7.7 | ||
| T | T s/c | 1 | 33.3 | 3.7 |
| Cortical madura | 2 | 66.7 | ||
| B-M | Bilineal B-M | 3 | 100 | 3.7 |
| NK | TLPNK | 1 | 100 | 1.2 |
| Total | — | 81 | — | 100 |
LA: leucemia aguda; s/c: sin clasificar; TLP: trastorno linfoproliferativo.
De acuerdo con su grado de diferenciación, las leucemias linfoblásticas agudas (LLA-B) se subclasificaron en pre-B I a IV7, correspondiendo la pre-B I a la pro-B, la pre-B II y III a la pre-B común o pre-B, y la pre-B IV a la B madura (en total 61/81; 75%).
- -
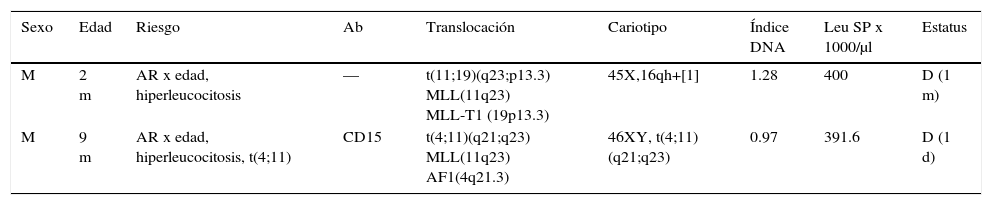
Pre-B I (pro-B; 2/61). Se identificaron dos casos de LLA pro-B, caracterizados por el inmunofenotipo CD34+, CD45int, nTdT+, CD19+, CD22+, HLA-DR+, CD38+, cCD79a+, CD10neg, CD20neg, cIgMneg (tabla 2). En esta serie, las LLA pro-B representaron el 3.3% de los casos de LLA-B y 2.9% del total de leucemias linfoblásticas. Ambos casos se presentaron en pacientes de sexo masculino menores de 1 año de edad; en los dos casos se detectó el reordenamiento 11q23 en el gen MLL, uno con la translocación t(11;19)(q23;p13.3) (MLL-T1) y el otro con la t(4;11)(q21;q23) (MLL-AF1), por lo que corresponden al subgrupo de la OMS leucemia/linfoma linfoblástico con rearreglo del gen MLL por translocación t(v;11q23); MLL (ICD-O-3; 9813/3). Estos casos presentaban hiperleucocitosis inicial (> 300,000/μl). Un caso mostró la expresión aberrante de CD15 y el otro hiperdiploidía (índice de DNA >1.16). Ambos casos fallecieron a corto plazo una vez diagnosticados.
Tabla 2.Leucemia pre-BI (pro-B)
Sexo Edad Riesgo Ab Translocación Cariotipo Índice DNA Leu SP x 1000/μl Estatus M 2 m AR x edad, hiperleucocitosis — t(11;19)(q23;p13.3) MLL(11q23)
MLL-T1 (19p13.3)45X,16qh+[1] 1.28 400 D (1 m) M 9 m AR x edad, hiperleucocitosis, t(4;11) CD15 t(4;11)(q21;q23) MLL(11q23)
AF1(4q21.3)46XY, t(4;11)(q21;q23) 0.97 391.6 D (1 d) Dos casos de 61 LLA-B (3.3%), (OMS ICD-O-3; 9813/3), con inmunofenotipo CD34+, CD45neg, nTdT+, CD19+, CD22+, cCD79a+, CD10neg, CD20neg, cIgMneg.
OMS ICD-O-3: Organización Mundial de la Salud, Clasificación Internacional de Enfermedades-Oncología-Versión 3; Ab: aberrante; Leu: leucocitos; SP: sangre periférica; M: masculino; AR: alto riesgo; D: defunción; M: masculino; m: meses; d: días.
- -
Pre-B II-III (pre-B común o pre-B; 57/61). De los 61 casos de LLA-B, 57 (93%) presentaron un perfil inmunofenotípico de leucemia pre-B II-III o LLA pre-B común (CD34+/-, CD45neg/int, nTdT+/-, CD19+, CD10+, CD20+/-, CD22+, HLA-DR+, cCD79a+/-, cIgM+/- κ o λ neg), desde lo más temprano (pre-B II; CD34+, cIgMneg) hasta lo más tardío en la diferenciación (pre-B III; CD34neg, cIgM+), sin llegar a la expresión de IgM en superficie. Estos casos se dividen entre aquellos con alteraciones genéticas (22/57; 39%) y sin ellas (35/57; 61%). Este subgrupo se designa a continuación como pre-B.
- •
Pre-B con alteraciones genéticas (22/57). Dentro de los casos con alteraciones genéticas se identificaron tres variantes: casos que presentan alteraciones estructurales (translocaciones y otras), con alteraciones numéricas (hiperdiploidía o hipodiploidía) y con ambas.
- ∘
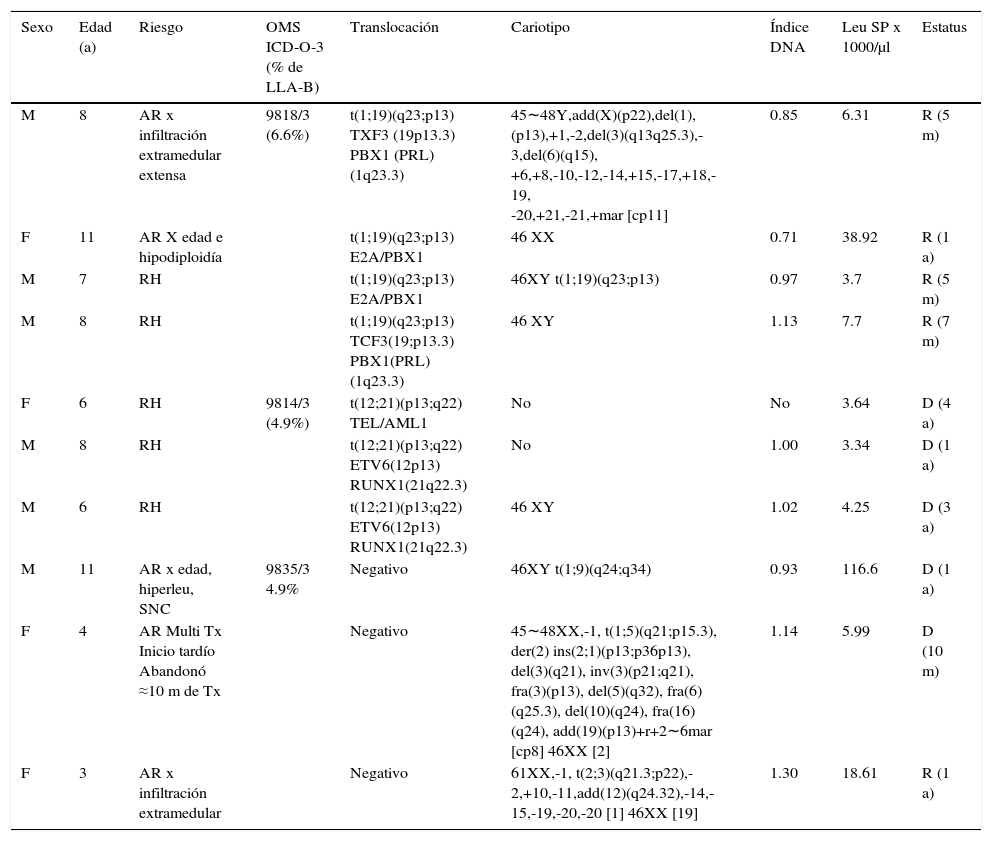
Translocaciones identificadas por RT-PCR o por cariotipo (10/22). Se detectaron seis casos de translocaciones por RT-PCR, tres por cariotipo y una por ambos métodos, para un total de 10 casos (17.5% de los casos pre-B; 16.4% de LLA-B; 14.7% de LLA) (tabla 3). La translocación más frecuente fue la t(1;19) (q23;p13) (subgrupo ICD-O-3; 9818/3 de la OMS), que se presentó en cuatro casos CD34 negativos (6.6% de LLA-B). Todos han tenido una evolución favorable y se encuentran en remisión después de 5 a 12 meses desde el diagnóstico, a pesar de presentar alto riesgo (AR) dos de ellos (uno por edad y uno por hipodiploidía y enfermedad extramedular extensa al inicio). Tres casos (5% de LLA-B) presentaron la translocación t(12;21)(p13;q22) (subgrupo ICD-O-3; 9814/3 de la OMS). Los tres fallecieron de 1 a 4 años desde el diagnóstico en recaída, a pesar de tener riesgo habitual de inicio y de haber respondido favorablemente al tratamiento inicial. Tres casos presentaron translocaciones no consideradas en la clasificación de la OMS: uno de ellos, que presentó la translocación t(1;9)(q24;q34), tuvo una evolución agresiva, con una segunda recaída en sistema nervioso central en menos de un año y falleció. Otro caso presentó la translocación t(1;5)(q21;p15.3), abandonó tratamiento a 10 meses del diagnóstico y también falleció. Un último caso de este grupo presentó la translocación t(2;3)(q21.3;p22) e hiperdiploidía, y ha evolucionado favorablemente. A un año del diagnóstico se encuentra en remisión a pesar de la infiltración extramedular extensa inicial.
Tabla 3.Leucemia pre-B, CD10 positivo (común) con translocaciones
Sexo Edad (a) Riesgo OMS ICD-O-3 (% de LLA-B) Translocación Cariotipo Índice DNA Leu SP x 1000/μl Estatus M 8 AR x infiltración extramedular extensa 9818/3
(6.6%)t(1;19)(q23;p13)
TXF3 (19p13.3)
PBX1 (PRL)(1q23.3)45∼48Y,add(X)(p22),del(1),(p13),+1,-2,del(3)(q13q25.3),-3,del(6)(q15),
+6,+8,-10,-12,-14,+15,-17,+18,-19,
-20,+21,-21,+mar [cp11]0.85 6.31 R (5 m) F 11 AR X edad e hipodiploidía t(1;19)(q23;p13) E2A/PBX1 46 XX 0.71 38.92 R (1 a) M 7 RH t(1;19)(q23;p13) E2A/PBX1 46XY t(1;19)(q23;p13) 0.97 3.7 R (5 m) M 8 RH t(1;19)(q23;p13) TCF3(19;p13.3) PBX1(PRL) (1q23.3) 46 XY 1.13 7.7 R (7 m) F 6 RH 9814/3
(4.9%)t(12;21)(p13;q22) TEL/AML1 No No 3.64 D (4 a) M 8 RH t(12;21)(p13;q22) ETV6(12p13) RUNX1(21q22.3) No 1.00 3.34 D (1 a) M 6 RH t(12;21)(p13;q22) ETV6(12p13) RUNX1(21q22.3) 46 XY 1.02 4.25 D (3 a) M 11 AR x edad, hiperleu, SNC 9835/3
4.9%Negativo 46XY t(1;9)(q24;q34) 0.93 116.6 D (1 a) F 4 AR Multi Tx Inicio tardío Abandonó ≈10 m de Tx Negativo 45∼48XX,-1, t(1;5)(q21;p15.3), der(2) ins(2;1)(p13;p36p13), del(3)(q21), inv(3)(p21;q21), fra(3)(p13), del(5)(q32), fra(6)(q25.3), del(10)(q24), fra(16)(q24), add(19)(p13)+r+2∼6mar [cp8] 46XX [2] 1.14 5.99 D (10 m) F 3 AR x infiltración extramedular Negativo 61XX,-1, t(2;3)(q21.3;p22),-2,+10,-11,add(12)(q24.32),-14,-15,-19,-20,-20 [1] 46XX [19] 1.30 18.61 R (1 a) Diez casos de 61 LLA-B (16.4%), con inmunofenotipo CD34+/-, CD45neg, nTdT+, CD19+, CD10+, CD20+/-, CD22+, cCD79a+/-, cIgM+/-.
OMS ICD-O-3: Organización Mundial de la Salud, Clasificación Internacional de Enfermedades-Oncología-Versión 3; Leu: leucocitos; SP: sangre periférica; M: masculino; F: femenino; AR: alto riesgo; SNC: sistema nervioso central; RH: riesgo habitual; Tx: tratamiento; R: remisión; D: defunción; Hiperleu: hiperleucocitosis; m: meses; a: años.
- ∘
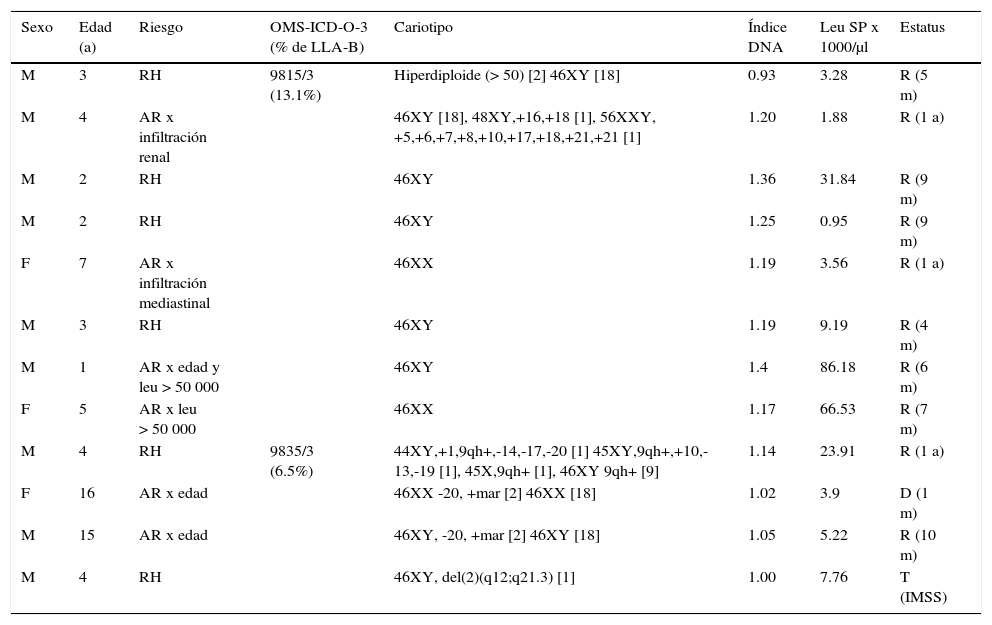
Alteraciones solamente del cariotipo (12/22). En la tabla 4 se presentan los 12 casos (21% de los casos pre-B; 20% de LLA-B; 15% de LLA) que presentaron solo alguna alteración cariotípica (se excluyeron de este grupo los casos que, además, presentaban translocaciones). En este grupo, seis casos se clasificaron inicialmente como de AR, por la edad o por enfermedad extensa con infiltración extramedular. Dos casos presentaron hiperleucocitosis de > 50 000/μl. Ocho de los casos presentaban hiperdiploidía (siete por índice de DNA >1.16 y uno por cariotipo) > 50 cromosomas (subgrupo ICD-O-3; 9815/3). Cuatro casos adicionales presentaban alteraciones cariotípicas diversas (ICD-O-3; 9835/3). A pesar de la asignación de AR (en seis pacientes) y de las alteraciones cariotípicas diversas, 10 de los 12 casos de este grupo se encontraban en remisión (de 4 meses a un año del diagnóstico). Un caso falleció al mes del diagnóstico y otro se transfirió a otra institución. Ningún caso de este grupo presentó hipodiploidía.
Tabla 4.Leucemia pre-B, CD10 positivo (común) con alteraciones cariotípicas
Sexo Edad (a) Riesgo OMS-ICD-O-3 (% de LLA-B) Cariotipo Índice DNA Leu SP x 1000/μl Estatus M 3 RH 9815/3
(13.1%)Hiperdiploide (> 50) [2] 46XY [18] 0.93 3.28 R (5 m) M 4 AR x infiltración renal 46XY [18], 48XY,+16,+18 [1], 56XXY, +5,+6,+7,+8,+10,+17,+18,+21,+21 [1] 1.20 1.88 R (1 a) M 2 RH 46XY 1.36 31.84 R (9 m) M 2 RH 46XY 1.25 0.95 R (9 m) F 7 AR x infiltración mediastinal 46XX 1.19 3.56 R (1 a) M 3 RH 46XY 1.19 9.19 R (4 m) M 1 AR x edad y leu > 50 000 46XY 1.4 86.18 R (6 m) F 5 AR x leu > 50 000 46XX 1.17 66.53 R (7 m) M 4 RH 9835/3
(6.5%)44XY,+1,9qh+,-14,-17,-20 [1] 45XY,9qh+,+10,-13,-19 [1], 45X,9qh+ [1], 46XY 9qh+ [9] 1.14 23.91 R (1 a) F 16 AR x edad 46XX -20, +mar [2] 46XX [18] 1.02 3.9 D (1 m) M 15 AR x edad 46XY, -20, +mar [2] 46XY [18] 1.05 5.22 R (10 m) M 4 RH 46XY, del(2)(q12;q21.3) [1] 1.00 7.76 T (IMSS) Doce casos de 61 LLA-B (19.6%) sin translocaciones detectadas, con inmunofenotipo CD34+, CD45neg, nTdT+, CD19+, CD10+, CD20+/-, CD22+, HLA-DR+, cCD79a+/-, cIgM+/-.
OMS ICD-O-3: Organización Mundial de la Salud, Clasificación Internacional de Enfermedades-Oncología-Versión 3; LLA: leucemia linfoblástica aguda; Leu: leucocitos; SP: sangre periférica; M: masculino; F: femenino; RH: riesgo habitual; AR: alto riesgo; R: remisión; D: defunción; T: traslado; m: meses; a: años; IMSS: Instituto Mexicano del Seguro Social.
- ∘
- •
Pre-B sin alteraciones genéticas (35/57). El grupo de casos de LLA pre-B donde no se identificaron alteraciones genéticas fue el más numeroso (sub-grupo ICD-O-3; 9811/3). De 35 casos (61% de LLA pre-B; 57% de LLA-B), 21 fueron de sexo masculino y 14 de femenino, edad promedio de 7.9 años; 18 fueron asignados inicialmente al grupo de AR o MAR (muy alto riesgo). Se encontró expresión aberrante de CD13 en cuatro pacientes, de CD36 en tres pacientes y de CD33 en un paciente. Siete de los 35 pacientes fallecieron de 1 mes a 4 años desde el diagnóstico. Veinticuatro continuaban en remisión de 2 a 19 meses desde el diagnóstico y cuatro habían abandonado el tratamiento.
- •
- -
LLA-B (madura; 2/61). En el grupo de las LLA-B se identificaron dos casos de LLA-B madura (CD34neg, CD45neg/int, nTdTneg, CD19+, CD10+/-, CD20+, CD22+, HLA-DR+, cCD79a+/-, cIgM+, κ+), caracterizados por la presencia de IgM en la superficie celular con cadenas ligeras en la población clonal neoplásica, con cadenas k (sub-grupo ICD-O-3; 9826/3). A los dos casos se les asignó un riesgo habitual; uno de ellos presentó hiperdiploidía (índice DNA 1.34) y cariotipo 46XY, 21ps+. No se encontraron translocaciones y ambos casos se encuentran en remisión de 3 a 9 meses desde el diagnóstico. No se determinó si existían alteraciones de c-Myc. Los casos se han tratado con el protocolo terapéutico de LLA.
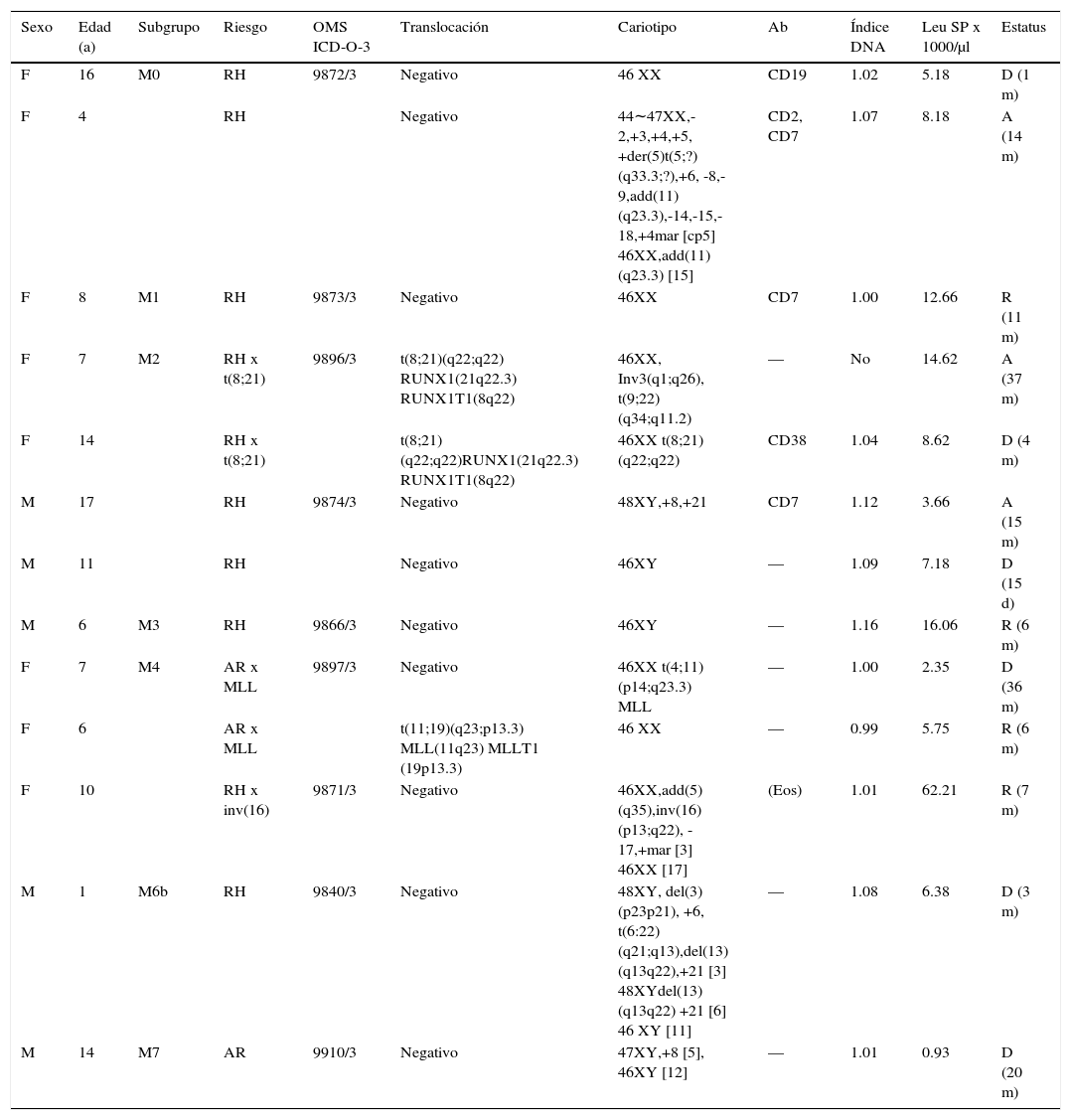
En la presente serie, el 16% de los casos (13/81) de LA atendidos en el HEP correspondieron a LMA. En estos casos, la relación por sexo fue 1.6:1 (femenino/masculino), y los pacientes fueron de edad promedio un poco mayor que los casos de LLA común (9.3 vs. 7.9 años) sin diferencia significativa (p > 0.05 determinada mediante la prueba t no pareada). En la tabla 5 se presenta la subclasificación de los casos mieloides con anotación de su asignación inicial de riesgo y su estado al momento del presente reporte. Nueve de los 13 casos (69%) presentaban translocaciones genéticas, alteraciones cariotípicas o ambas. De estos casos, fallecieron (6/9) o abandonaron el tratamiento en recaída (3/9) de 15 días a 37 meses desde el diagnóstico, sin una relación clara con el perfil genético; siete de ellos con riesgo habitual inicial. Solo cuatro de los 13 casos estaban en remisión de 6 a 11 meses desde el diagnóstico, uno de ellos con AR inicial. Los dos casos (ambos M2) que presentaban la translocación (t8;21)(q22;q22) evolucionaron desfavorablemente. Uno falleció a los 4 meses y el segundo evolucionó a partir de leucemia mieloide crónica, cromosoma Filadelfia positivo —con la translocación t(9;22)(q34;q11.2) BCR-ABL1—, para la cual recibió imatinib y nilotinib más hidroxiurea con buenas respuestas temporales; abandonó tratamiento en recaída M2 a los 37 meses de evolución. El caso M4 con alteración del gen MLL (11q23.3) por la translocación t(4;11)(p14;q23.3) falleció a los 36 meses del diagnóstico. El otro caso M4 con alteración del gen MLL debida a la translocación t(11;19)(q23;p13.3) continuaba en remisión a 6 meses del diagnóstico. El tercer caso M4 se presentó con eosinofilia y la inversión inv(p13;q22); fue el único caso de la serie con hiperleucocitosis de > 50 000/μl y se encontraba en remisión a 7 meses del diagnóstico. El caso M6b con la translocación t(6:22)(q21;q13) y otras alteraciones cariotípicas falleció a tres meses del diagnóstico. Por último, un caso M7 persistió con bicitopenia severa (anemia y leucopenia) y trombocitosis moderada por varios meses, sin recibir tratamiento antineoplásico. Finalmente presentó una crisis mieloblástica y falleció. La médula ósea y la sangre periférica mostraron blastos con un fenotipo M7 (CD34+, CD36+, CD61+).
Leucemia mieloide
| Sexo | Edad (a) | Subgrupo | Riesgo | OMS ICD-O-3 | Translocación | Cariotipo | Ab | Índice DNA | Leu SP x 1000/μl | Estatus |
|---|---|---|---|---|---|---|---|---|---|---|
| F | 16 | M0 | RH | 9872/3 | Negativo | 46 XX | CD19 | 1.02 | 5.18 | D (1 m) |
| F | 4 | RH | Negativo | 44∼47XX,-2,+3,+4,+5, +der(5)t(5;?)(q33.3;?),+6, -8,-9,add(11) (q23.3),-14,-15,-18,+4mar [cp5] 46XX,add(11)(q23.3) [15] | CD2, CD7 | 1.07 | 8.18 | A (14 m) | ||
| F | 8 | M1 | RH | 9873/3 | Negativo | 46XX | CD7 | 1.00 | 12.66 | R (11 m) |
| F | 7 | M2 | RH x t(8;21) | 9896/3 | t(8;21)(q22;q22) RUNX1(21q22.3) RUNX1T1(8q22) | 46XX, Inv3(q1;q26), t(9;22)(q34;q11.2) | — | No | 14.62 | A (37 m) |
| F | 14 | RH x t(8;21) | t(8;21)(q22;q22)RUNX1(21q22.3) RUNX1T1(8q22) | 46XX t(8;21)(q22;q22) | CD38 | 1.04 | 8.62 | D (4 m) | ||
| M | 17 | RH | 9874/3 | Negativo | 48XY,+8,+21 | CD7 | 1.12 | 3.66 | A (15 m) | |
| M | 11 | RH | Negativo | 46XY | — | 1.09 | 7.18 | D (15 d) | ||
| M | 6 | M3 | RH | 9866/3 | Negativo | 46XY | — | 1.16 | 16.06 | R (6 m) |
| F | 7 | M4 | AR x MLL | 9897/3 | Negativo | 46XX t(4;11)(p14;q23.3) MLL | — | 1.00 | 2.35 | D (36 m) |
| F | 6 | AR x MLL | t(11;19)(q23;p13.3) MLL(11q23) MLLT1 (19p13.3) | 46 XX | — | 0.99 | 5.75 | R (6 m) | ||
| F | 10 | RH x inv(16) | 9871/3 | Negativo | 46XX,add(5)(q35),inv(16)(p13;q22), -17,+mar [3] 46XX [17] | (Eos) | 1.01 | 62.21 | R (7 m) | |
| M | 1 | M6b | RH | 9840/3 | Negativo | 48XY, del(3)(p23p21), +6, t(6:22)(q21;q13),del(13)(q13q22),+21 [3] 48XYdel(13)(q13q22) +21 [6] 46 XY [11] | — | 1.08 | 6.38 | D (3 m) |
| M | 14 | M7 | AR | 9910/3 | Negativo | 47XY,+8 [5], 46XY [12] | — | 1.01 | 0.93 | D (20 m) |
Trece casos de 81 LA (81%). OMS ICD-O-3: Organización Mundial de la Salud, Clasificación Internacional de Enfermedades-Oncología-Versión 3; Ab: aberrante; Leu: leucocitos; SP: sangre periférica; M: masculino; F: femenino; AR: alto riesgo; A: abandono; R: remisión; D: defunción d: días; m: meses; a: años.
Se observaron tres casos de leucemia aguda de fenotipo mixto (LAFM) para el 3.7% de las LA (3/81; subgrupo ICD-O-3; 9805/3). Dos de los casos fueron de línea B y monocítica; uno presentó hipodiploidía (índice DNA 0.8) e hiperleucocitosis (> 100 000/μl) y se encuentra en inducción de remisión. El otro caso tenía la translocación t(9;22)(q34;q13) (OMS 9806/3) y abandonó tratamiento al mes de diagnóstico. El tercer caso, de B y M (sin definir) se encuentra en remisión a 58 meses del diagnóstico. Los tres son verdaderos casos bilineales (dos poblaciones de blastos caracterizables), sin expresión bifenotípica en una línea celular o expresión de aberrantes de acuerdo con el criterio del EGIL.
3.2.4Leucemia linfoblástica aguda TEl grupo de LLA T incluyó tres casos (3/81; subgrupo ICD-O-3; 9837/3). En dos de ellos se estableció la subclasificación inmunofenotípica “cortical madura” (CD45neg, CD34neg, nTdT+, CD1a+/-, CD2+, cCD3+, CD3+, CD4+, CD5+, CD7+, CD8+, TCRα/β+/-, TCRγ/δ+/-). El otro caso no fue subclasificado. Los tres casos cursaron con hiperleucocitosis (> 100 000/μl). En un caso se identificó hipodiploidía y se acompañó de masa mediastinal; en otro se identificó la alteración TAL1 (T-cell Acute Leukemia-1) del STIL (1p32). Un caso con más de 400 000 leucocitos/μl en sangre periférica no aceptó el tratamiento el día del diagnóstico y fue dado de alta. Los dos restantes continuaban en remisión a los 6 y 27 meses del diagnóstico.
3.2.5Transtorno linfoproliferativo de células NK (TLPC NK)Se identificó un caso (1/81; ICD-O-3 9831/3) de TLPC NK (CD45int, CD34+, CD10neg, CD19neg, CD13neg, MPOneg, CD2neg, CD3neg, CD4neg, CD5neg, CD8neg, CD56/16+, CD58+ con expresión aberrante de CD7) en un menor de 2 años, que evolucionó por 2 meses con fiebre, dolores articulares y óseos intensos, palidez y manifestaciones hemorrágicas, sin visceromegalias, con pancitopenia periférica y con la translocación t(11;22)(p11.2;q12.3). El familiar solicitó el alta voluntaria al mes del ingreso.
3.3EtnicidadTodos los pacientes reportados eran nativos del estado de Chiapas. El 60.5% (49/81) pertenecía a familias indígenas, de las cuales el 55% (27) habitaban en localidades rurales de alto o muy alto grado de marginación. El resto perteneció a familias con un grado variable de mestizaje que radicaban en la periferia de zonas urbanas y suburbanas, según lo reportado en un estudio socioeconómico.
4DiscusiónLos resultados de 81 casos estudiados detalladamente desde varios puntos de vista (morfológico, inmunofenotípico y genotípico), con información de su evolución y destino a corto plazo, permiten reconocer los tipos y la diversidad de LA que se ha observado en pacientes —mayoritariamente indígenas— atendidos en el HEP del estado de Chiapas y proporcionan información integral indispensable relativa a las particularidades inherentes a dicha enfermedad en la población atendida.
De acuerdo con el objetivo primario de este trabajo, y considerando únicamente la línea celular leucémica, es evidente que las poblaciones pediátricas autóctonas del estado de Chiapas atendidas en el HEP muestran un patrón similar al que se observa en otras partes del país10,11, a pesar de su relativo aislamiento y alto grado de consanguinidad. En la mayor parte de los casos se observan afectados los linfocitos B, seguidos por las leucemias mieloides, las linfocíticas T, casos aislados de bifenotípicas y otras variedades, como los NK (tabla 1).
Dentro de las leucemias B, las leucemias pro-B que se presentan en el primer año de la vida (leucemia infantil) se asocian con alteraciones del gen MLL y con frecuente expresión aberrante de CD15, tal como se ha reportado en otros sitios. Estas leucemias presentan un comportamiento agresivo para las cuales se ha reportado un mal pronóstico12–14. Estos hechos han sugerido que la leucemia pro-B infantil es una enfermedad determinada por alteraciones genéticas leucemogénicas, producidas durante la gestación del paciente en un precursor linfo-monocítico temprano, y por tanto diferente de la leucemia pre-B adquirida a otras edades12,13. El 3.3% (2/61) de las LLA-B encontrado en esta población es comparable al 1.2% (3/251) de leucemias B reportado por Bekker-Méndez y colaboradores en niños mexicanos (mayoritariamente mestizos) atendidos en la Ciudad de México15. Los reportes en otros países y otros centros mexicanos varían ampliamente (desde 0 a 65.4%). Por su naturaleza y mal pronóstico, el abordaje de estos casos podría incluir el planteamiento temprano de trasplante de médula, tratamientos más agresivos y estrategias derivadas del conocimiento del mecanismo molecular íntimo, consecuencia de las alteraciones del gen MLL, como el uso de inhibidores de la TAL1 (metil-transferasa que interactúa con proteínas transcripcionales asociadas al gen MLL, inhibiendo la apoptosis de las células malignas), entre otras acciones16.
A diferencia del subgrupo anterior, donde la alteración del gen MLL es definitoria, en las leucemias pre-B las alteraciones genéticas cuantitativas y cualitativas pueden o no estar presentes. Sin embargo, cuando están presentes, dan lugar a subgrupos específicos con características clínicas particulares. En esta serie, las presentaron el 36% de los casos de LLA B (22/61); diez de las 22 (16.3% de las LLA B) consistieron en translocaciones con o sin alteraciones cariotípicas. Este porcentaje es muy similar al 17.7% reportado por Bekker-Méndez y colaboradores15. La translocación más frecuente (6.6% de LLA B) fue la t(1;19)(q23;p13) (ICD-O-3; 9818/3), que fusiona el gen PBX1 del cromosoma 1 con el gen TCF3 (E2A) del cromosoma 19, dando lugar a la proteína nuclear de transcripción 5’E2A/3’PBX1, que activa constitutivamente la transcripción de genes regulados por la familia de genes PBX17. Aun cuando la presencia de esta translocación se asociaba originalmente con mal pronóstico, con los esquemas terapéuticos actuales el pronóstico se considera favorable18,19. Esta perspectiva coincide con la evolución de los casos presentados, ya que los cuatro casos con esta translocación se encontraban en remisión de 5 meses a 1 año desde el diagnóstico, uno de ellos a pesar de presentar hipodiploidía y el otro a pesar de tener enfermedad extramedular extensa. En la serie de Bekker-Méndez y colaboradores se presentó en el 7.1% de las LLA B y 19 de 20 pacientes estaban vivos después de 1 año del diagnóstico15.
La segunda translocación más frecuente en esta serie fue la t(12;21)(p13;q22) (ICD-O-3; 9814/3), que da lugar a la fusión de los genes TEL (ETV6) del cromosoma 12 y el AML1 en el cromosoma 21, y a la subsecuente producción de la proteína quimérica TEL/AML1 con función activadora de transcripción de varios genes específicos de la hematopoyesis20. La presentaron tres casos (5.0%) y, contrariamente a lo encontrado en la literatura occidental donde se reporta como de buen pronóstico21,22, los tres casos, aunque remitieron rápidamente, fallecieron en recaída de 1 a 4 años desde el diagnóstico. Este resultado difiere de lo observado por Bekker-Méndez y colaboradores, ya que en dicha serie se presentó en el 7.4% de los casos de LLA B, y solamente un paciente (1/21) falleció en el primer año. De confirmarse esta diferencia con el análisis de más casos, se establecería una particularidad local importante.
Se observaron tres casos con sendas translocaciones: la t(1;9)(q24;q34), la t(1;5)(q21;p15.3) y la t(2;3)(q21.3;p22). La primera ha sido reportada previamente en LLA23; fusiona el gen RCSD1 del cromosoma 1, que codifica la proteína CapZIP (relacionada con la formación del citoesqueleto celular), con el gen ABL1 del cromosoma 9, que codifica una tirosina cinasa específica. Sin embargo, no existen datos suficientes en la literatura en cuanto a su comportamiento clínico. El paciente con esta translocación falleció 10 meses después del diagnóstico. La segunda translocación, t(1;5)(q21;p15.3), no se ha reportado en leucemias. Se detectó, entre otras alteraciones genéticas, en un caso multitratado de una paciente de sexo femenino de 4 años de edad, que falleció a los 10 meses del diagnóstico. El tercer caso presentaba además hiperdiploidía, y se encuentra en remisión al año del diagnóstico. No se localizaron en la literatura reportes de esta translocación en casos de LLA.
Destaca la ausencia de casos con la translocación t(9:22)(q34;q11) en este subgrupo, ya que es una de las alteraciones que se presenta en otras series nacionales y extranjeras15,24,25. Esta translocación define los casos de LLA cromosoma Filadelfia positivos, y por tanto candidatos a recibir tratamiento adyuvante con los inhibidores de la tirosina cinasa disponibles actualmente para el tratamiento de la leucemia mieloide crónica clásica. Aún con este recurso, estos casos tienen mal pronóstico. Por ello, la translocación t(9;22) ha sido incorporada como criterio de asignación al grupo de MAR por el Children's Oncology Group (COG)22.
Además de las translocaciones, se encuentran alteraciones cariotípicas dentro de las cuales destaca la hiperdiploidía inespecífica sin translocación concurrente que corresponde al grupo ICD-O-3; 9815/3 de la OMS, ya que estuvo presente en 8 de los 61 casos de LLA B (13.1%). En congruencia con la información internacional disponible26, los resultados en este estudio sugieren que, en ausencia de una translocación específica, la hiperdiploidía es un factor dominante de evolución favorable, ya que independientemente de la clasificación de riesgo inicial por la edad, cifra de leucocitos y grado de infiltración de las células malignas, los ocho casos con hiperdiploidía se encontraban en remisión de 4 meses a 1 año desde el diagnóstico. Cuatro casos adicionales presentaron alteraciones cariotípicas diversas: dos se encontraban en remisión, uno falleció y el otro se transfirió a otra institución (tabla 4).
El subgrupo de leucemias pre-B sin alteraciones genéticas constituye, al igual que en otras series, el grupo más numeroso. Estos casos se han comportado como en otros centros: el 20% falleció (de 1 mes a 4 años desde el diagnóstico), el 11.4% abandonó el tratamiento y el 68.5% continúa en remisión (de 2 a 19 meses desde el diagnóstico).
Los dos casos identificados como leucemia B madura se consideran actualmente como una variante del linfoma de Burkitt (OMS 9826/3). Habitualmente presentan una alteración del gen c-Myc, secundaria a la translocación t(8;14)(q24;q32), determinando la expresión constitutiva del gen Myc8. El tratamiento de estos casos como linfoma Burkitt ha cambiado radicalmente su pronóstico; actualmente se recomienda una combinación de quimio e inmunoterapia27. Los casos de la presente serie no se estudiaron para c-Myc ni se identificaron translocaciones. Se trataron de acuerdo con el protocolo nacional para leucemias, y su evolución ha sido favorable.
La frecuencia relativa de LMA observada en el presente estudio es similar a lo que se ha observado en todo el país11,28 y en la población hispana en el extranjero29, lo que sugiere una relación étnica con esta variedad. La clasificación en subgrupos atomiza los casos, ya que la OMS incluye cuando menos 20 subgrupos o variantes de LMA asociados o no con translocaciones específicas9. La distribución de estos casos en los subgrupos principales de la FAB-OMS es un tanto homogénea ya que, con excepción de dos casos M0 y dos M2 con la translocación t(8;21), los demás subgrupos aportaron un caso de cada uno de los subgrupos observados (tabla 5). En adultos, ha sido posible establecer tres grupos pronóstico relacionados con translocaciones específicas30. En pacientes pediátricos existe mayor diversidad de opiniones que generalmente incorporan la respuesta inicial al tratamiento31.
La escasez de casos en esta serie impide una valoración significativa de su comportamiento. En general, al margen de la subclasificación, la evolución de los casos de LMA ha sido mala. Solamente con la acumulación de más casos podrá establecerse una idea de la evolución de las diferentes variedades y su comparación con la información internacional31.
Las leucemias agudas de fenotipo mixto (LAFM; OMS 9805-9809/3)8, antes bilineales o bifenotípicas, constituyeron del 2 al 5% de las leucemias, al igual que en otras partes del mundo32. Los tres casos que se presentaron fueron B-M (monocíticas), pero también pueden presentarse T-M y B-T. No existe un protocolo nacional específico para el tratamiento de esta variedad y, en general, no existen diferencias de comportamiento con las LA de una sola línea celular.
Las LLA T se presentaron con incidencia comparable a las LAFM, a diferencia de lo que se observa en países orientales, donde llega a constituir el 15% de las LA. Clásicamente se consideran de mal pronóstico, ya que frecuentemente se acompañan de hiperleucocitosis de > 100 000 blastos/μl, masa mediastinal, lisis tumoral, infiltración del SNC y complicaciones renales secundarias. Sin embargo, con los tratamientos actuales, se han logrado sobrevidas a 5 años de más del 60% de los casos33. La evolución de los casos presentados ha sido muy favorable, con buenas respuestas a pesar de hipodiploidía y enfermedad extensa.
En la medida en que se avanza en el conocimiento inmunológico y molecular de las leucemias, y se aplica para su identificación, clasificación y subclasificación, se van configurando subconjuntos clínicos dentro de una misma línea celular afectada de comportamiento y pronóstico característicos. La existencia de estos subgrupos plantea la posibilidad de considerar a la LA como un verdadero síndrome, causado por diversas enfermedades moleculares únicas. Sin embargo, dada la relativamente baja frecuencia de alteraciones moleculares puntuales distintivas (por ejemplo, translocaciones ≈17%), también es posible que la LA sea una enfermedad única, cuya base molecular permanece oculta, y que los subgrupos identificados representen alteraciones genéticas y epigenéticas secundarias a la presunta alteración primaria común.
Las poblaciones pediátricas autóctonas del estado de Chiapas que son atendidas en el HEP, a pesar de su relativo aislamiento y alto grado de consanguinidad, mostraron una proporción del tipo primario de leucemias similar al que se observa en otras partes del país. Otros hechos similares son la frecuencia, las alteraciones moleculares y la mala evolución de la leucemia B infantil, el desenlace habitualmente fatal y diversidad de las leucemias mieloides; la buena evolución de casos con la translocación t(1;19)(q23;p13) en las leucemias B y el carácter favorable de la hiperdiploidía en todos los casos.
También existen diferencias específicas en esta serie, de lo local con lo nacional: la mala evolución de las leucemias B con la translocación t(12;21)(p13;q22) (TEL/AML1) comparado con el buen pronóstico en otros sitios; la ausencia de la translocación t(9:22)(q34;q11); la ausencia de casos de trisomía 21 a pesar de que el síndrome de Down es relativamente frecuente en la población local; la detección de translocaciones no reportadas previamente en leucemia, como la t(1;5)(q21;p15.3) y la t(2;3)(q21.3;p22); la buena evolución de los casos con leucemia/linfoma de Burkitt tratados con el Protocolo Nacional para leucemias; la detección de casos complejos bilineales y mieloides de comportamiento único (por ejemplo, M7 con evolución subaguda) y la buena evolución de las leucemias T a pesar de hiperleucocitosis extremas e hipodiploidía.
Es muy probable que, en un futuro cercano, el estudio del genoma completo en cada paciente aumente las diferencias locales de las nacionales e internacionales, por lo que habrá que continuar el análisis comparativo de los casos y de sus particularidades locales26,34.
Este estudio pretende servir como soporte inicial para análisis posteriores enfocados a consolidar o rectificar los hallazgos encontrados, con el objetivo último de personalizar los tratamientos para la leucemia que dependerían de las características individuales, tanto clínicas como citopatológicas, con asignación de riesgo sustentada en datos objetivos locales35.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciamientoRecursos propios: Hospital de Especialidades Pediátricas, Chiapas, México; Fundación “Gonzalo Río Arronte I.A.P.”; Fundación “Casa de la Amistad para niños con Cáncer I.A.P.”.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores desean expresar su agradecimiento al M. en C. de la Computación Joaquín Octavio Docelis Burguete y al ingeniero Ronald Martínez Gómez por su invaluable apoyo informático y bioestadístico, así como a las Fundaciones “Gonzalo Río Arronte I.A.P.” y “Casa de la Amistad para niños con Cáncer I.A.P.” por la donación del citómetro de flujo y reactivos que permitieron la realización del presente trabajo.