




Las malformaciones congénitas vertebrales y costales concomitantes comprenden un grupo heterogéneo de enfermedades denominadas disostosis espondilocostal. Tienen en común la alteración del desarrollo o morfología de las estructuras vertebrales y de la caja torácica con una expresividad variable: desde la deformidad leve sin consecuencias funcionales hasta lesiones que amenazan la vida. Se presenta el caso de una niña con disostosis espondilocostal y colangitis aguda.
Caso clínicoPaciente de sexo femenino de 13 meses de edad con desnutrición severa y antecedente de hidrocefalia y mielomeningocele quien ingresa al servicio de Urgencias por presentar dificultad respiratoria progresiva y fiebre. En la evaluación se encontraron malformaciones costovertebrales y colangitis aguda.
ConclusionesLas anormalidades costales complejas consisten en malformaciones de la pared torácica sin un patrón determinado y son extremadamente raras. Cuando se presentan al mismo tiempo que las malformaciones vertebrales, puede considerarse como síndrome de disostosis espondilocostal ligado a herencia autosómica recesiva. El diagnóstico es clínico-radiográfico. La identificación de la disostosis espondilocostal y las complicaciones relacionadas con sus causas genético-moleculares implican un reto para el pediatra y el equipo multidisciplinario que los trata a lo largo de su vida.
Congenital malformations of the chest wall comprise a heterogeneous group of diseases denominated spondylocostal dysostosis. They have in common developmental abnormalities in the morphology of the structures of the chest and vertebrae with a broad characterization: from mild deformity without functional consequences to life-threatening injuries. We present the case of a girl with spondylocostal dysostosis and acute cholangitis.
Clinical caseA 13-month-old girl with severe malnutrition, history of hydrocephalus and myelomeningocele at birth was admitted in the emergency pediatric room with fever and progressive respiratory distress. Clinical assessment revealed ribs and vertebral malformations and acute cholangitis.
ConclusionsComplex rib abnormalities consist in deformities of the chest wall, which do not have a specific pattern and are extremely rare. When they are associated with myelomeningocele and hydrocephalus they may be considered as autosomal recessive inheritance spondylocostal dysostosis. The diagnosis is established by clinical assessment and X-rays. Spondylocostal dysostosis identification and complications related to their genetic and molecular causes are still a challenge for clinical pediatricians and the multidisciplinary medical team who treats these patients throughout lifetime.
Las malformaciones costales congénitas de la pared del tórax se clasifican en cinco tipos: tipo I, cartilaginosas (pectus excavatum, pectus carinatum); tipo II, costales (simples: únicas, dobles, combinadas; complejas: fusiones, sindromáticas); tipo III, costocondrales (síndrome de Poland, toracópagos); tipo IV, esternales (fisura esternal); tipo V, clavículo-escapulares (claviculares, escapulares, combinadas). Pueden formar parte de síndromes, como disostosis espondilocostal y disostosis espondilotorácica que se caracterizan por anomalías costales y vertebrales con o sin defectos del tubo neural y malformaciones extraesqueléticas, o pueden presentarse como defectos aislados1. En la disostosis espondilocostal se encuentran múltiples defectos de segmentación vertebral y anormalidades costales (fusión, reducción en número, desalineación).
Clínicamente se caracteriza por un tronco corto en proporción a la talla, cuello corto y escoliosis. El diagnóstico se basa en los hallazgos radiográficos. Los subtipos se definen por la identificación de dos alelos mutantes transmitidos por herencia autosómica recesiva en cualquiera de los cuatro genes afectados que se conocen (DLL3, MESP2, LFNG y HES7)2, cuya alteración afecta la vía de señalización Notch, la cual resulta crítica para la coordinación de este proceso; o herencia autosómica dominante por alteraciones en la activación de la transcripción de la proteína TBX6, probablemente debidas a haploinsuficiencia3.
2Caso clínicoPaciente de sexo femenino de 13 meses de edad con habitus externo ectomórfico, macrocefalia por hidrocefalia con reservorio de válvula de derivación ventrículo-peritoneal derecha funcional, la cual fue colocada a los 22 días de vida extrauterina (cicatriz quirúrgica de 2cm en fosa iliaca derecha). Presentó mielomeningocele, corregido al nacimiento (cicatriz quirúrgica de 8cm en región sacrococcígea). Producto de la segunda gesta obtenida a las 36.6 semanas de gestación por cesárea; embarazo sin control prenatal. Hermana mayor de 5 años y padres referidos sanos que negaron consanguinidad, toxicomanías, enfermedades crónicas o degenerativas, exposición a tóxicos ambientales o presencia de lesiones similares en otros familiares. Al ingreso al servicio de Urgencias el peso fue de 7kg (percentil < 3 para la edad de acuerdo con las tablas de la Organización Mundial de la Salud), la talla de 63cm (percentil < 3 para la edad) y el perímetro cefálico de 55cm (percentil > 97 para la edad). Tono muscular respetado, trofismo disminuido, deambulación con ayuda. Los signos vitales fueron frecuencia cardíaca 131 latidos/min, 42 respiraciones/min, temperatura axilar 38.5°C, saturación de oxígeno 97% por oximetría de pulso, presión arterial 94/49mmHg.
Ingresó por fiebre, dificultad respiratoria progresiva, hepatomegalia de 4cm; asimetría en la excursión torácica secundaria a hipomotilidad, hemitórax derecho, depresión leve de pared torácica por palpación y durante la auscultación, protrusión discreta durante el llanto. No se observaron otros elementos clínico-patológicos (tabla 1).
Resumen de los elementos clínicos y radiográficos de la paciente
| Antecedentes |
| Hidrocefalia |
| Defecto del tubo neural (mielomeningocele corregido) |
| Exploración física |
| Macrocefalia con hidrocefalia |
| Peso bajo para la edad, normal para la talla |
| Talla baja para la edad (restricción del segmento torácico) |
| Retraso en el desarrollo del lenguaje, se para y camina con ayuda |
| Fiebre |
| Dificultad respiratoria |
| Asimetría torácica derecha |
| Hepatomegalia |
| Elementos radiográficos |
| Agenesia de primera costilla derecha |
| Primera costilla izquierda hipoplásica |
| Inserción aberrante de ambas clavículas |
| Sexto y séptimo arcos costales fusionados en hemitórax derecho |
| Vértebras T6 y T7 en mariposa |
| Gastromegalia |
| Enfermedad actual |
| Colangitis |
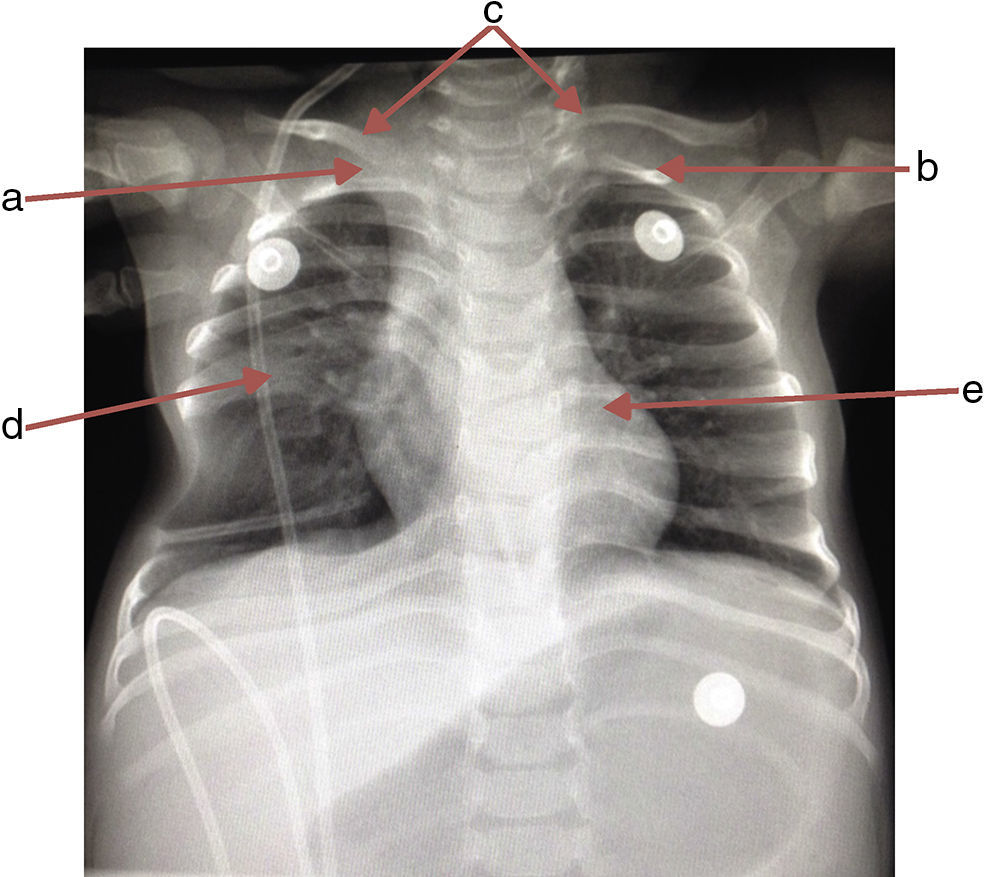
Se solicitó radiografía de tórax en la que se observó la ausencia de la primera costilla derecha y la primera costilla izquierda hipoplásica, lo que propiciaba la inserción aberrante de las clavículas. El sexto y séptimo arcos costales fusionados en hemitórax derecho, desplazados hacia arriba; los inferiores, hacia abajo formando un espacio que realza la transparencia del pulmón, y las vértebras T6 y T7 en mariposa (fig. 1).
, hipoplasia en la primera costilla izquierda (b), inserción aberrante de ambas clavículas (c), sexto y séptimo arcos costales fusionados en hemitórax derecho (d) y vértebras T6 y T7 en mariposa (e).")
Radiografía anteroposterior de tórax donde se observa la ausencia de la primera costilla derecha (a), hipoplasia en la primera costilla izquierda (b), inserción aberrante de ambas clavículas (c), sexto y séptimo arcos costales fusionados en hemitórax derecho (d) y vértebras T6 y T7 en mariposa (e).
Se realizó ecografía en la que se observaron hígado y vías biliares; se encontró colédoco con dilatación fusiforme de 1.9cm cerca de su unión con el conducto de Wirsung. Se sospechó colangitis y se solicitó evaluación por Infectología Pediátrica, quienes sugirieron iniciar tratamiento con piperacilina-tazobactam (previa realización de hemocultivo). Se solicitó intervención multidisciplinaria con los servicios de Nutrición, Cirugía Torácica y Neumología, quienes recomendaron tratamiento conservador con citas para evaluar el desarrollo torácico y pulmonar cada 3 meses. El servicio de Fisiología Pulmonar instruyó a la madre sobre medidas de higiene pulmonar; 24h más tarde fue trasladada al servicio de Cirugía Pediátrica, donde se realizó una colangiografía laparoscópica y se confirmó colangitis.
Se inició el proceso encaminado a la recuperación nutricional dentro del hospital. Catorce días después egresó asintomática y se citó para seguimiento.
3DiscusiónEntre las semanas 3-5 de gestación, el mesodermo (entre el endodermo y ectodermo, ambos lados de la notocorda) se diferencia en somitas y da lugar al esclerotomo (procesos costales y vertebrales), el miotomo (musculatura esquelética del tronco) y el dermatomo (capas profundas de la piel y del tejido celular subcutáneo)1,4. La formación de somitas a partir de su tejido precursor, el mesodermo presomita, se encuentra controlada en su totalidad a nivel molecular por la interacción de varías vías de señalización, que incluyen las vías FGF, Wnt y Notch. En este proceso, la vía de señalización Notch se activa en el mesodermo presomita en pulsos regulares, lo que conduce a la activación periódica de los genes LFNG y HES75. Cualquier trastorno durante esta etapa de embriogénesis puede resultar en defectos vertebrales, costales y abdominales. También se ha implicado al gen Hox en la malformación costal6.
El término disostosis espondilocostal se emplea para describir una variedad de hallazgos radiográficos que incluyen múltiples defectos en la segmentación vertebral, generalmente contiguos, acompañados de estructuras costales desalineadas, fusionadas o ausentes5,7–9.
Las malformaciones costales pueden presentarse con insuficiencia respiratoria al nacimiento, pasar inadvertidas o progresar a falla respiratoria durante el desarrollo; por ello, el diagnóstico puede retardarse hasta la vida adulta. El presente caso corresponde a una malformación costal tipo II (3.2% del total de las malformaciones costales). Se asocia con alteraciones anatómicas poco comunes; cada una constituye una variedad única cuyo pronóstico e implicaciones diagnósticas y de tratamiento son individuales. Los diagnósticos diferenciales de las malformaciones costales incluyen el síndrome de Poland10, el síndrome cerebrocostomandibular, el síndrome de Edwards11,12, el síndrome de VACTERL-H13, el síndrome lumbocostoverterbral14,15, la disostosis espondilotorácica (síndrome de Jarcho-Levin16,17, síndrome de Cassamassima-Morton-Nance18 con elevada mortalidad por insuficiencia respiratoria) (tabla 2) y disostosis espondilocostal1–4.
Comparación entre diagnósticos diferenciales y caso índice
| Diagnóstico diferencial | Año de publicación por primera vez | Características clínicas | Caso índice |
|---|---|---|---|
| Síndrome de Poland10 Herencia autosómica dominante | 1841 | • Aplasia unilateral de los fascículos costoesternales del músculo pectoral mayor • Falta de pectoral muscular menor • Hipoplasia mamaria y del pezón • Braquisindactilia unilateral • Deformidad de la pared torácica • Hipoplasia costal | x x x x x √ |
| Síndrome cerebro-costomandibular Herencia tanto autosómica dominante como recesiva | 1966 | • Microcefalia • Anomalías costales bilateral asimétricas • Micrognatia | x √ x |
| Síndrome de Edwards11,12 Herencia autosómica recesiva ligada al cromosoma X | 1960 | • Anomalías costales (clavículas afiladas, costillas estrechas) • Anomalías vertebrales • Mielomeningocele • Microcefalia • Occipucio prominente • Paladar ojival • Micrognatia | x √ √ x x x x |
| Síndrome de VACTERL-H13 Herencia autosómica recesiva | 1972 | • Debe tener tres o más anomalías para el diagnóstico: | |
| • Defectos vertebrales | √ | ||
| • Defectos cardiacos | x | ||
| • Defectos renales | x | ||
| • Extremidades en pulgares o radio | x | ||
| • Atresia anal | x | ||
| • Fístula tráqueo-esofágica | x | ||
| • Atresia esofágica o duodenal | x | ||
| • Hidrocefalia | √ | ||
| Síndrome lumbocostovertebral14,15 Madres diabéticas, exposición al ácido lisérgico, probable herencia autosómica recesiva | 1972 | • Anomalías costovertebrales y de la musculatura del tronco: • Hemivértebras • Ausencia de costillas • Disrafia espinal • Mielomeningocele anterior • Hipoplasia de la pared abdominal anterior (hernia lumbosacra) • Hepatopatía • Artrogriposis • Cardiopatía congénita • Agenesia renal | √ √ √ x x x x x x |
| Síndrome de Cassamassima-Morton-Nance (OMIM 271520)18 Afectación genes Pax 1 y Pax 9. Herencia autosómica recesiva, mutaciones in novo y rearreglos cromosómicos | 1981 | • Defectos de segmentación • Tórax corto • Anomalías costales (fusión) • Vertebrales asimétricas • Hemivértebras, fusión vertebral • Talla baja, tronco corto • Espina bífida/mielomeningocele • Dificultad o insuficiencia respiratoria • Escoliosis, atresia anal, malformación genitourinaria/anal (característico) • Rasgos dismórficos cráneo-faciales, cuello corto y con poca movilidad • Anomalías cardíacas | √ √ √ √ √ √ √ √ x x x |
| • Frente amplia, dolicocefalia, puente nasal ancho, narinas antevertidas, boca en carpa (ocasionales) | x | ||
| • Déficit intelectual (rasgo ocasional) | x | ||
| Síndrome de Jarcho-Levin16,17 Herencia autosómica dominante | 1938 | • Talla baja (por alteraciones espinales) a expensas de segmento superior corto | √ |
| • Anomalías vertebrales (principal característica) | √ | ||
| • Anomalías costales | √ | ||
| • Defectos del tubo neural (mielomengingocele) | √ | ||
| • Hidrocefalia | √ | ||
| • Dificultad/insuficiencia respiratoria | √ | ||
| • Infecciones respiratorias recurrentes | √ | ||
| • Cardiovasculares (defectos septales) | x | ||
| • Hernia diafragmática | x | ||
| • Genitourinarios | x | ||
| • Divertículo de Meckel | x | ||
| • Úvula bífida | x |
√: signo presente; x: signo ausente.
En 1968, Rimoin y colaboradores propusieron el término de displasia espondilocostal para las malformaciones localizadas en la columna vertebral y costillas. En 1991, Karnes y colaboradores, basados en hallazgos radiográficos, redefinieron el síndrome Jarcho-Levin en dos tipos: disostosis espondilotorácica y disostosis espondilocostal. Mortier y colaboradores, en 1996, dividieron los defectos de segmentación múltiple en tres tipos: síndrome de Jarcho-Levin (tórax simétrico con aspecto de “cangrejo”), disostosis espondilotorácica y disostosis espondilocostal; todos con posibles malformaciones extraesqueléticas7,17,19,20.
Actualmente, el diagnóstico de disostosis espondilocostal es clínico-radiográfico; sin embargo, existe gran variedad de fenotipos de imágenes descritos, los cuales han sido empleados para denominar las anormalidades costales y vertebrales. Esto ha generado confusión en la nomenclatura. Por tal motivo, el Consorcio Internacional para Anomalías Vertebrales y Escoliosis (ICVAS, por sus siglas en inglés) propuso un algoritmo para la investigación ontogénica que facilita la comparación y estratificación de los defectos espinales (curvatura, longitud), vertebrales (normal, segmentación única o múltiple y morfología), de la caja torácica (simetría, asimetría, tamaño, forma) y costales (simetría, asimetría, número, fusión)2,4,8.
Las malformaciones costales acompañadas de defectos del tubo neural y talla baja secundaria a alteraciones espinales son compatibles con disostosis espondilocostal, que es un desorden genético raro con una prevalencia de 0.25/10,000 nacidos vivos16. Pertenece a un grupo de enfermedades hereditarias caracterizadas por un defecto en la segmentación vertebral múltiple (hemivértebras, agenesia, en mariposa e hipoplásicas) con alteraciones costales (fusiones, agenesia, desalineación) asociadas con defectos del tubo neural, como la malformación de Arnold-Chiari e hidrocefalia. Cada malformación neurológica que acompaña al síndrome es uno de sus componentes; por ejemplo, la disgenesia del cuerpo calloso, holoproscencefalia, mielomeningocele (lumbosacro, torácico o lumbar) y holoproscencefalia. Además, pueden existir anormalidades renales, genito-urinarias, gastrointestinales, de extremidades y cardíacas congénitas.
Para confirmar y establecer el diagnóstico en un individuo afectado se deben buscar otras anomalías esqueléticas en las radiografías, realizar ecosonografías (cardíaca, abdominal y renal), y posteriormente determinar si los hallazgos clínicos y radiológicos son consistentes con alguno de los trastornos incluidos en el diagnóstico diferencial. Además, se debe investigar la historia familiar, en especial los casos de individuos afectados o de consanguinidad de los padres.
Una vez que el diagnóstico de disostosis espondilocostal se ha establecido, se emplea el fenotipo radiográfico para la determinación de los posibles genes implicados.
La disostosis espondilocostal por causa genética se ha clasificado en dos grupos: en el primero se encuentran las formas severas de disostosis espondilocostal, con malformación de 10 o más vértebras, generalmente ligada a la transmisión autosómica recesiva con penetración completa. Incluye las mutaciones en los genes DLL3 (SCDI; OMIM 277300), MESP2 (SCD2; OMIM 608681), LFNG (SCD3; OMIM 609813) y HES7 (SCD4; OMIM 613686). También se encuentra en este grupo el síndrome disostosis espondilotorácica, fenotípicamente distinto, causado por mutaciones en el gen MESP2. En la forma autosómica dominante de disostosis espondilocostal solamente se encuentran afectadas algunas vértebras; se ha demostrado que es debido a una haploinsuficiencia con penetración variable e incluye mutaciones en TBX6. El síndrome Klippel-Feil es causado por mutaciones en GDF6 (KFSI; OMIM 118100) o GDF3 (KFS3; OMIM 613702) y la escoliosis congénita es causada por mutaciones en MESP2 o HES72–4,8. Es conveniente señalar la somatogénesis de los genes HES y HEY, ya que están involucrados en la regulación de la neurogénesis, vasculogénesis, cardiogénesis y cáncer.
Los cuatro tipos de disostosis espondilocostal por herencia autosómica recesiva presentan fenotipos radiográficos distintivos. Se requiere de mejores evidencias para determinar si el genotipo se correlaciona con los fenotipos 3 y 4. Las características clínicas comprenden tronco corto en proporción a la talla, cuello corto, escoliosis leve generalmente no progresiva, y defectos de segmentación costo-vertebral, entre otras.
Disostosis espondilocostal 1 (asociada con el gen DLL3). Comprende los cuatro criterios diagnósticos más un patrón irregular de osificación de los cuerpos vertebrales en la edad prenatal y en la primera infancia. Cada cuerpo presenta una forma redonda u ovoide con límites lisos (signo de la playa de piedras). Existe predominio de afectación torácica. Se ha reportado consanguinidad en el 75% de los casos.
Disostosis espondilocostal 2 (asociada con MESP2). Todos los segmentos vertebrales muestran interrupciones en su forma; las vértebras lumbares son las más afectadas. Se ha reportado consanguinidad.
Disostosis espondilocostal 3 (asociada con LFNG). Acortamiento de la columna vertebral más severo que en los tipos 1 y 2. Todos los cuerpos vertebrales tienen defectos graves de segmentación. Las anomalías costales son similares a los tipos 1 y 2. Se asemeja a enfermedades de transmisión sexual. Ha sido reportada en una sola familia.
Disostosis espondilocostal 4 (asociada con HES7). Se asemeja a la disostosis espondilotorácica con anomalías de segmentación vertebral más severas. Los pedículos vertebrales son relativamente prominentes (signo del tranvía) en comparación con el tipo 1. Se ha informado en dos familias del sur de Europa. Afectación del cromosoma 17p13.1 (fenotipo MIM 613686) por mutación de HES7 (gen/locus MIM 608059) lo cual se ha asociado con defectos en la lateralidad y formación del tubo neural4,5.
Las disostosis espondilotorácicas, a pesar de sus similitudes con las disostosis espondilocostales autosómicas recesivas, presentan características fenotípicas individuales (tabla 3)21. Se han descrito en personas con enfermedades de transmisión sexual y la afectación del gen MESP2. Además, han sido ampliamente descritas por Cornier y colaboradores en individuos puertorriqueños de ascendencia española22. Este fenotipo se conoce actualmente como síndrome Jarcho-Levin, y cuando se acompaña de ano imperforado, malformaciones genitourinarias y otras extraesqueléticas, se le denomina síndrome de Cassamassima-Morton-Nance20.
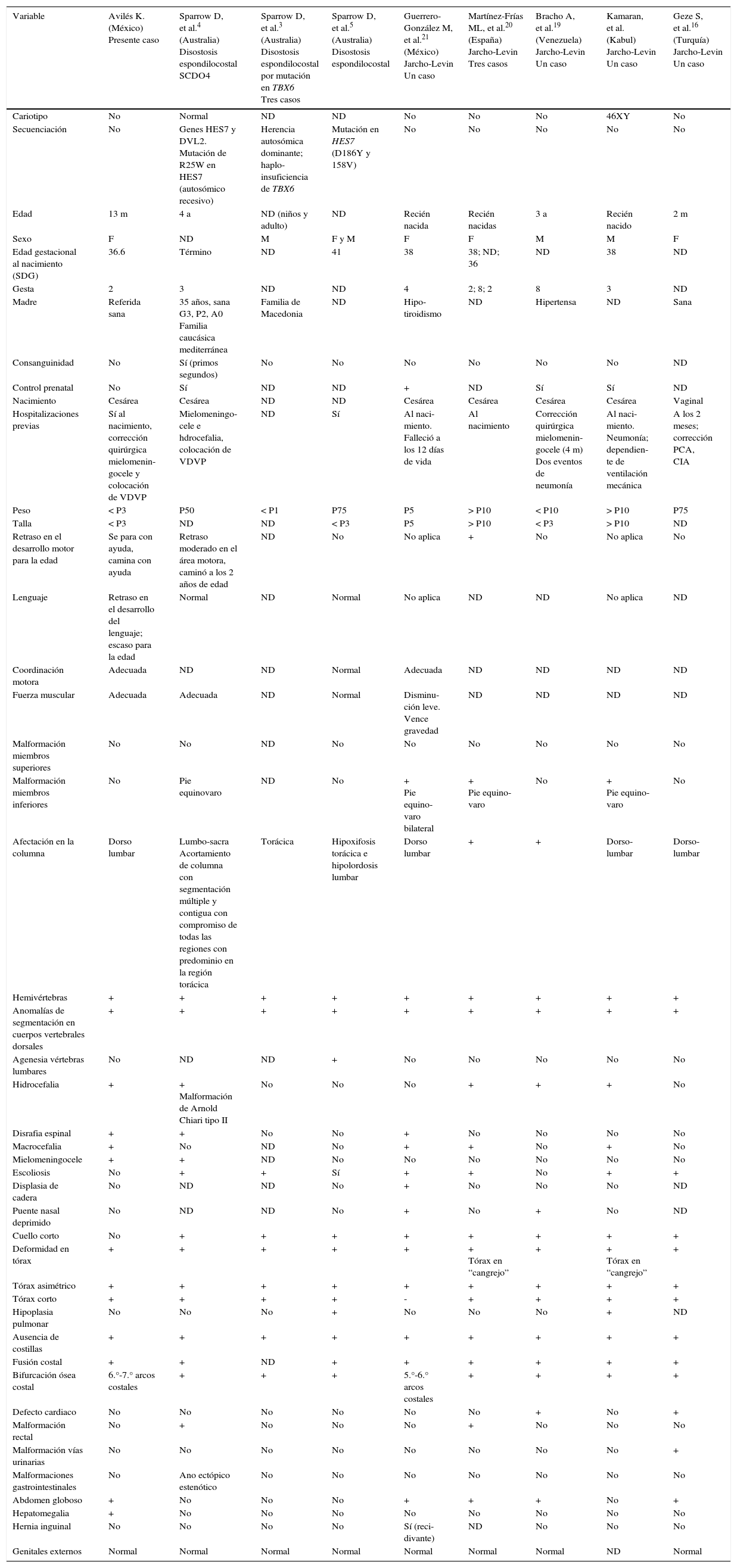
Comparación del caso índice con otros casos reportados en la literatura con malformaciones costo-vertebrales por disostosis espondilocostal y disostosis espondilotorácica
| Variable | Avilés K. (México) Presente caso | Sparrow D, et al.4 (Australia) Disostosis espondilocostal SCDO4 | Sparrow D, et al.3 (Australia) Disostosis espondilocostal por mutación en TBX6 Tres casos | Sparrow D, et al.5 (Australia) Disostosis espondilocostal | Guerrero-González M, et al.21 (México) Jarcho-Levin Un caso | Martínez-Frías ML, et al.20 (España) Jarcho-Levin Tres casos | Bracho A, et al.19 (Venezuela) Jarcho-Levin Un caso | Kamaran, et al. (Kabul) Jarcho-Levin Un caso | Geze S, et al.16 (Turquía) Jarcho-Levin Un caso |
|---|---|---|---|---|---|---|---|---|---|
| Cariotipo | No | Normal | ND | ND | No | No | No | 46XY | No |
| Secuenciación | No | Genes HES7 y DVL2. Mutación de R25W en HES7 (autosómico recesivo) | Herencia autosómica dominante; haplo-insuficiencia de TBX6 | Mutación en HES7 (D186Y y 158V) | No | No | No | No | No |
| Edad | 13 m | 4 a | ND (niños y adulto) | ND | Recién nacida | Recién nacidas | 3 a | Recién nacido | 2 m |
| Sexo | F | ND | M | F y M | F | F | M | M | F |
| Edad gestacional al nacimiento (SDG) | 36.6 | Término | ND | 41 | 38 | 38; ND; 36 | ND | 38 | ND |
| Gesta | 2 | 3 | ND | ND | 4 | 2; 8; 2 | 8 | 3 | ND |
| Madre | Referida sana | 35 años, sana G3, P2, A0 Familia caucásica mediterránea | Familia de Macedonia | ND | Hipo-tiroidismo | ND | Hipertensa | ND | Sana |
| Consanguinidad | No | Sí (primos segundos) | No | No | No | No | No | No | ND |
| Control prenatal | No | Sí | ND | ND | + | ND | Sí | Sí | ND |
| Nacimiento | Cesárea | Cesárea | ND | ND | Cesárea | Cesárea | Cesárea | Cesárea | Vaginal |
| Hospitalizaciones previas | Sí al nacimiento, corrección quirúrgica mielomenin-gocele y colocación de VDVP | Mielomeningo-cele e hdrocefalia, colocación de VDVP | ND | Sí | Al naci-miento. Falleció a los 12 días de vida | Al nacimiento | Corrección quirúrgica mielomenin-gocele (4 m) Dos eventos de neumonía | Al naci-miento. Neumonía; dependien-te de ventilación mecánica | A los 2 meses; corrección PCA, CIA |
| Peso | < P3 | P50 | < P1 | P75 | P5 | > P10 | < P10 | > P10 | P75 |
| Talla | < P3 | ND | ND | < P3 | P5 | > P10 | < P3 | > P10 | ND |
| Retraso en el desarrollo motor para la edad | Se para con ayuda, camina con ayuda | Retraso moderado en el área motora, caminó a los 2 años de edad | ND | No | No aplica | + | No | No aplica | No |
| Lenguaje | Retraso en el desarrollo del lenguaje; escaso para la edad | Normal | ND | Normal | No aplica | ND | ND | No aplica | ND |
| Coordinación motora | Adecuada | ND | ND | Normal | Adecuada | ND | ND | ND | ND |
| Fuerza muscular | Adecuada | Adecuada | ND | Normal | Disminu-ción leve. Vence gravedad | ND | ND | ND | ND |
| Malformación miembros superiores | No | No | ND | No | No | No | No | No | No |
| Malformación miembros inferiores | No | Pie equinovaro | ND | No | + Pie equino-varo bilateral | + Pie equino-varo | No | + Pie equino-varo | No |
| Afectación en la columna | Dorso lumbar | Lumbo-sacra Acortamiento de columna con segmentación múltiple y contigua con compromiso de todas las regiones con predominio en la región torácica | Torácica | Hipoxifosis torácica e hipolordosis lumbar | Dorso lumbar | + | + | Dorso-lumbar | Dorso-lumbar |
| Hemivértebras | + | + | + | + | + | + | + | + | + |
| Anomalías de segmentación en cuerpos vertebrales dorsales | + | + | + | + | + | + | + | + | + |
| Agenesia vértebras lumbares | No | ND | ND | + | No | No | No | No | No |
| Hidrocefalia | + | + Malformación de Arnold Chiari tipo II | No | No | No | + | + | + | No |
| Disrafia espinal | + | + | No | No | + | No | No | No | No |
| Macrocefalia | + | No | ND | No | + | + | No | + | No |
| Mielomeningocele | + | + | ND | No | No | No | No | No | No |
| Escoliosis | No | + | + | Sí | + | + | No | + | + |
| Displasia de cadera | No | ND | ND | No | + | No | No | No | ND |
| Puente nasal deprimido | No | ND | ND | No | + | No | + | No | ND |
| Cuello corto | No | + | + | + | + | + | + | + | + |
| Deformidad en tórax | + | + | + | + | + | + Tórax en “cangrejo” | + | + Tórax en “cangrejo” | + |
| Tórax asimétrico | + | + | + | + | + | + | + | + | + |
| Tórax corto | + | + | + | + | - | + | + | + | + |
| Hipoplasia pulmonar | No | No | No | + | No | No | No | + | ND |
| Ausencia de costillas | + | + | + | + | + | + | + | + | + |
| Fusión costal | + | + | ND | + | + | + | + | + | + |
| Bifurcación ósea costal | 6.°-7.° arcos costales | + | + | + | 5.°-6.° arcos costales | + | + | + | + |
| Defecto cardiaco | No | No | No | No | No | No | + | No | + |
| Malformación rectal | No | + | No | No | No | + | No | No | No |
| Malformación vías urinarias | No | No | No | No | No | No | No | No | + |
| Malformaciones gastrointestinales | No | Ano ectópico estenótico | No | No | No | No | No | No | No |
| Abdomen globoso | + | No | No | No | + | + | + | No | + |
| Hepatomegalia | + | No | No | No | No | No | No | No | No |
| Hernia inguinal | No | No | No | No | Sí (reci-divante) | ND | No | No | No |
| Genitales externos | Normal | Normal | Normal | Normal | Normal | Normal | Normal | ND | Normal |
m: meses; a: años; SDG: semanas de gestación; VDVP: válvula derivativa ventrículo peritoneal; P: percentil para la edad; ND: no descrito; +/–: elemento variable presente/ausente; PCA: persistencia de conducto arterioso; CIA: comunicación interauricular.
Con respecto a las infecciones agudas del tracto biliar en ausencia de atresia de vías biliares, se sabe que son infrecuentes en la edad pediátrica (0.13%-0.22%)23. La condición aguda y grave por inflamación e infección en el ducto biliar se define como colangitis y se caracteriza por hepatalgia, fiebre e ictericia (triada de Charcot). La colangitis aguda es un proceso sistémico con alta mortalidad, por lo que el tratamiento médico resulta urgente24. Este es el primer caso reportado del que se tiene conocimiento de una niña con disostosis espondilocostal y colangitis aguda; esta última resulta un factor de riesgo que complica la condición preexistente.
En los casos con disostosis espondilocostal, Teli y colaboradores han reportado que el tratamiento conservador mediante fisioterapia torácica da buenos resultados para incrementar la sobrevida; la complicación más temida es la insuficiencia respiratoria. El tratamiento quirúrgico se reserva para niños con ausencia de respuesta: aquellos que requieren de estabilización de la caja torácica o deformidades espinales, como la escoliosis progresiva9.
Después de la evaluación integral del caso, la clasificación del fenotipo radiográfico de acuerdo con las malformaciones vertebrales y costales, el antecedente de mielomeningocele e hidrocefalia más una amplia revisión de la literatura, se determinó que el caso presentado correspondía a una disostosis espondilcostal tipo 4. No se encontró una descripción para explicar la presencia de colangitis bacteriana aguda en una niña sin factores de riesgo descritos, como la malformación de vías biliares.
Las malformaciones costales complejas son infrecuentes; si se acompañan de anormalidades vertebrales y defectos del tubo neural, se debe considerar el síndrome de disostosis espondilocostal. El diagnóstico de la variedad corresponde al fenotipo radiográfico. La importancia del diagnóstico radica en establecer el tratamiento para prevenir la insuficiencia respiratoria, otras complicaciones intratorácicas, del desarrollo vertebral y las extraesqueléticas, y posteriormente el seguimiento multidisciplinario. Sin embargo, la clasificación para la disostosis espondilocostal y sus variantes es aún complicada, por lo que resulta un reto para el pediatra y el equipo multidisciplinario que les atiende a lo largo de su vida.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciamientoNinguno.
Conflicto de interesesLa autora declara no tener ningún conflicto de intereses.
Al doctor Eloy López-Marure, médico radiólogo pediatra adscrito al servicio de Radiología e Imagen del Hospital Civil Juan I. Menchaca, Guadalajara, Jalisco, México; y al doctor Hugo Gutiérrez-González, residente de Pediatría del Hospital Civil Fray Antonio Alcalde, Guadalajara, Jalisco, México.