



Paciente masculino de 5 años de edad con fiebre, dolor abdominal e ictericia.
1.1Antecedentes heredofamiliaresMadre de 27 años de edad, escolaridad primaria terminada, católica, niega toxicomanías, unión libre, aparentemente sana. Padre de 35 años de edad, albañil, católico, aparentemente sano. Cuenta con carga genética para diabetes mellitus y hepatopatía.
1.2Antecedentes personales no patológicosOriginario y residente del D.F., casa propia con servicios básicos. Convivía con un perro.
Alimentación. Integrado a la dieta familiar, con dieta indicada por gastronutrición.
Desarrollo psicomotor. Acorde para la edad.
Inmunizaciones. Refiere esquema de vacunación completo (en trasplantes se refieren vacunas faltantes para poder enlistarse).
1.3Antecedentes perinatales y patológicosProducto de la segunda gestación, con adecuado control prenatal, USG normales, ingesta de multivitamínicos. Embarazo normoevolutivo, obtenido de término, peso de 2,700g. Se desconocen talla y Apgar. Sin complicaciones perinatales.
Paciente conocido por el Hospital Infantil de México Federico Gómez desde enero de 2007, por hepatopatía crónica, con antecedentes de ictericia desde la primera semana de vida.
4 de julio de 2007 (patología). Se hizo el diagnóstico de obstrucción parcial al flujo biliar.
3 de septiembre de 2008. Se trató como hepatitis autoinmune e inició esteroides. Posteriormente presentó elevación de transaminasas a pesar del tratamiento; los esteroides se disminuyeron paulatinamente y se suspendieron. IgG elevada, LKM (anticuerpos antimicrosomales tipo 1 de hígado y riñón) 2+, anticuerpos antinucleares 1:80 moteado fino, antirribosomal 9.4.
25 de junio de 2009. Inició tratamiento con prednisona durante 2meses. En agosto se disminuyó la dosis de esteroides hasta suspenderlos. No hubo mejoría en los niveles de aminotransferasas.
8 de septiembre de 2010. Se realizó biopsia hepática sugestiva de diagnóstico de fibrosis hepática.
1.4Padecimiento actualPresentó fiebre de 72h de evolución de inicio súbito, no progresivo, en dos picos cuantificados de 38°C, con duración de 35min. Tuvo dolor abdominal de 48h de evolución de inicio insidioso, progresivo, difuso, intensidad moderada, no se relacionó con la ingesta de alimentos. Presentó evacuaciones disminuidas en consistencia de 24h de evolución, con inicio súbito progresivo en seis ocasiones, líquidas, verdosas, fétidas, sin moco ni sangre.
Se agregó ictericia de 48h de evolución de inició súbito, progresivo, cefalocaudal.
Presentó alteración del ciclo sueño/vigilia de 24h de evolución; la madre lo refirió como falta de sueño durante la noche.
Exploración física. Tinte ictérico con mucosas secas, pupilas isocóricas hiporrefléxicas. Abdomen blando, depresible, sin datos de irritación peritoneal, distendido, globoso a expensas de líquido de ascitis, peristalsis no audible, hepatoesplenomegalia y red venosa colateral.
Las extremidades eran hipotróficas, el llenado capilar de 2”, con buena coloración y temperatura. Neurológicamente, irritable, con Glasgow de 8, sin datos de focalizaciones ni paresias.
Laboratorios. Leucocitos 9,900; N 58%; L 15%; B 22%; Hb 9.8; Hto 29; PLT 49,000; FA 558; BUN 36; calcio 7.7; gasometrías: pH 7.31, pCO2 68.5, Sat 90.6%, HCO3 13.8, amonio 233, TP 18.4, INR 1.45, TTP 33.
Diagnósticos de ingreso
- -
Desnutrición aguda grave
- -
Cirrosis hepática criptogénica
- -
Encefalopatía hepática grado iii
- -
Anemia normocítica normocrómica
- -
Hiperuricemia
- -
Trombocitopenia
- -
Choque séptico
- -
Probable colangitis
Evolución. Se refirió llenado capilar retardado hasta de 4” asociado con el deterioro del estado neurológico (Glasgow de 8), por lo que se inició fase iii de ventilación. Posteriormente se refirió hipotensión y taquicardia que requirió carga de cristaloides; no hubo respuesta adecuada. Se inició administración de dobutamina y se consultó al servicio de Infectología por probable sepsis abdominal. Se sugirió cobertura empírica para sepsis abdominal con ampicilina, cefotaxima y metronidazol.
Fue valorado también por los servicios de Trasplante y la Unidad de Terapia Intensiva Pediátrica. Se encontró pendiente el estudio de VIH y la aplicación de vacuna de varicela para ser listado ante el Centro Nacional de Trasplantes (CENATRA). Presentó anuria de 3h de evolución, con alto riesgo para falla orgánica múltiple, además de hematoquecia, por lo que se decidió transfundir concentrado eritrocitario, plasma fresco congelado y plaquetas. Continuó con sangrado de tubo digestivo bajo. Se cuantificaron hasta 300ml. Se administraron 4 unidades de concentrados plaquetarios, 225ml de concentrados eritrocitarios y 150ml de plasma fresco congelado. Fue valorado por el servicio de Cirugía por sangrado, quienes aconsejaron seguir con manejo a base de líquidos y transfusión. Continuó con hemorragia y se inició manejo con octreotide en infusión. Presentó hipertensión arterial y bradicardia, por lo que se disminuyó la dosis a 0.5μg/kg. Era dependiente de oxígeno con puntas nasales a 0.5l/min. En el transcurso del día presentó cuadro de hematemesis (50ml), por lo que se colocó sonda nasogástrica con derivación y lavado gástrico. Posteriormente presentó hematoquecia (700ml/4h). Hb 8g/dl, TP 19.6 y TTP 28.8. Se dejó nuevamente en ayuno, reiniciando transfusión de hemoderivados. Presentó hipotensión y disminución de uresis (0.94ml/kg/h). Se aumentó la infusión de octreotide a 1μg/kg/h.
4 de mayo de 2011. Hemodinámicamente estable, sin sangrado, dependiente de oxígeno en puntas nasales. Hemoglobina de 0.3, por lo que se decidió reiniciar la vía oral.
5 de mayo de 2011. En sesión conjunta con Gastroenterología y Nutrición se decidió realizar escleroterapia por várices grado iii-iv, por el alto riesgo de sangrado. La biometría hemática de control indicó Hb de 8.8; la cifra previa fue de 9.4.
6 de mayo de 2011. Se realizó la ligadura de várices y escleroterapia. En la endoscopia se observó el esófago con cordón incipiente en el tercio medio, y a nivel del tercio distal se apreciaron cuatro cordones varicosos grado iii-iv, así como una huella de esclerosis previa. En el estómago no había huellas de sangrado activo. Se procedió a ligadura de várices; se logró en dos cordones varicosos. Posteriormente, se realizó escleroterapia en tres cordones varicosos con polidocanol al 1.5%. Se suspendió el octreotide. Presentó hipertensión secundaria a hipervolemia; se administró diurético de asa.
10 de mayo de 2011. Presentó dos evacuaciones con hematoquecia y pobre tolerancia a la vía oral. La hemoglobina fue de 9.8, las plaquetas de 55,000; se transfundió concentrado plaquetario y plasma fresco congelado. Se reinició administración de octreotide. Además se observó irritabilidad, por lo que se indicó amonio.
13 de mayo de 2011. Presentó evento súbito de deterioro ventilatorio, datos de encefalopatía y saturación de 60%, arterial. Las radiografías de tórax mostraron infiltrado bilateral macronodular, y presentó gingivorragia y epistaxis. Neurológicamente evidenció deterioro con alteración de sueño/vigilia, irritabilidad y Glasgow de 11. Se decidió intubarlo. Posteriormente tuvo un paro cardiorrespiratorio, por lo que se administraron aminas y reanimación avanzada. Durante el evento presentó datos de hemorragia masiva de tubo digestivo y pulmón. Neurológicamente, con Glasgow 3. TP 19, TTP 35, fibrinógeno 197, Hb 13, plaquetas 133,000, AST 98, ALT 35, BI 22, BD17. Dos horas después presentó otro paro cardiorrespiratorio con duración de 9min. Se comentó con los padres el estado grave del paciente y el mal pronóstico a corto plazo, y ellos decidieron que no se realizaran maniobras de reanimación avanzada.
Diagnósticos de egreso
- -
Fibrosis hepática congénita variedad colangítica
- -
Hipertensión portal
- -
Várices esofágicas grado iii-iv en tercio distal, várices fúndicas grandes y gastropatía hipertensiva
- -
Desnutrición crónica agudizada de intensidad moderada
- -
Encefalopatía hepática grado iii
- -
Choque séptico remitido
- -
Insuficiencia renal aguda prerrenal remitida
- -
Probable colangitis
- -
Hemorragia pulmonar masiva
El caso que nos compete trata de un preescolar masculino de 5 años de edad con múltiples antecedentes de importancia, entre los cuales destaca el contar con familiares directos con patología hepática y de la vía biliar. Presentó colestasis neonatal aparentemente resuelta en el segundo mes de vida, sin factores que pudieran considerarse causa directa de esta.
Con base en los signos y síntomas de la historia clínica, se integraron los siguientes diagnósticos sindromáticos:
- 1.
Síndrome colestásico. Con base en ictericia que se presentó desde la primera semana de vida, y asociado con niveles de bilirrubina directa por encima de 1mg/dl, con la total<5; no asociado a acolia o hipocolia.
- 2.
Síndrome de hipertensión portal. Manifestado por ascitis, red venosa colateral y esplenomegalia, además de várices esofágicas y fúndicas de acuerdo con lo que reportaron los hallazgos endoscópicos.
- 3.
Síndrome de respuesta inflamatoria sistémica. Por la presencia de fiebre, hipotensión y bandemia.
- 4.
Síndrome diarreico agudo con deshidratación. Por la presencia de seis evacuaciones disminuidas en consistencia, de 24h de evolución, asociadas con la sequedad de las mucosas.
- 5.
Síndrome hemorragíparo. Manifestado por epistaxis de repetición, hematuria, gingivorragia, hematemesis y melena.
- 6.
Síndrome encefálico. Con base en la alteración del ciclo sueño/vigilia, irritabilidad, así como disminución del estado de alerta; sin datos de focalización.
- 7.
Síndrome de insuficiencia respiratoria aguda. Con base en taquipnea de instauración súbita, disminución de la oxemia e infiltrado bilateral paroxístico.
Con los diagnósticos sindromáticos previamente mencionados se integraron los siguientes diagnósticos nosológicos:
- 1.
Hepatopatía crónica. Por presentar elevación persistente de aminotrasferasas durante un periodo superior a 6 meses, asociada con colestasis y elevación de la gamma-glutamil traspeptidasa.
- 2.
Insuficiencia hepática. Por presentar alteraciones como coagulopatía, hiperamonemia, hipoglucemia, hipoalbuminemia secundaria sumada a la encefalopatía hepática que se describió.
- 3.
Choque séptico. Ante los datos de respuesta inflamatoria sistémica, con foco gastrointestinal y pulmonar, refractario a volumen que requirió la administración de aminas vasoactivas.
- 4.
Choque hipovolémico. Por presentar sangrado a nivel gingival, nasal, de tubo digestivo alto y bajo asociado con coagulopatía, con repercusión hemodinámica, sumado a hemorragia pulmonar masiva, que se refiere por sangrado activo asociado al deterioro ventilatorio súbito que presentó el paciente.
Se trata de un preescolar con hepatopatía crónica de base, cuya etiología no fue clara. Al momento de ser referido a este instituto, el paciente era un lactante menor que presentaba ya un estadio muy avanzado de afectación hepática evidenciada por los datos clínicos, y apoyada por la biopsia en la que se observó cirrosis biliar al año y 6 meses de edad. Se desconoce si existió infiltrado inflamatorio.
Al avanzar la cirrosis, la afectación en la arquitectura hepática condicionó alteraciones en la vasculatura así como en la estructura biliar. Con ello se perpetuó el daño hepático, aun cuando el agente agresor inicial había cedido.
El hecho de presentar cirrosis biliar desde el primer año de vida habla de un daño hepático extenso de tipo fibrótico, comúnmente asociado con un patrón obstructivo, y marcada colestasis. El hecho de presentar evacuaciones pigmentadas, así como la visibilidad de la vesícula biliar por ecografía, no descarta la posibilidad de una etiología anatómica, pero la aleja considerablemente.
Es importante destacar que el paciente debió de ser estudiado desde la segunda semana de vida, en la que persistió con ictericia, haberse documentado colestasis neonatal y, sin importar la presencia o ausencia de pigmento en las evacuaciones, se debió haber realizado un abordaje diagnóstico completo. Ante la complejidad que integra el abordaje y consideraciones diagnósticas, es importante evaluar las pistas con las que se cuenta.
La cirrosis hepática puede tener múltiples causas. En el caso de este paciente, la visceromegalia permite sospechar enfermedades por depósito, particularmente depósito lisosomal. De la misma forma, una de las causas preponderantes de la cirrosis biliar es la fibrosis quística, la cual, aparentemente, fue descartada.
Al continuar con las consideraciones diagnósticas, se observó que en este paciente las causas infecciosas se volvieron sobresalientes ante el antecedente de colestasis neonatal. En el resumen de la historia clínica no se encontró evidencia de haber descartado citomegalovirus, Epstein Barr, toxoplasma, rubéola, VIH, que en ocasiones cursan con afección hepática. Además, no se descartó la posibilidad de que se trate de una infección viral recurrente. Incluso, se debió haber inmunizado para hepatitis A, hepatitis B y varicela ante el riesgo de adquisición de nuevas infecciones, para así evitar un daño hepático mayor.
Los tóxicos de importancia clínica son otra entidad a descartar en este tipo de pacientes ante las prácticas culturales de consumo de tés, hongos y algunas otras sustancias que pueden afectar el hígado, a tal grado de causar una falla hepática fulminante, no presente en el paciente.
Otra posibilidad diagnóstica es la enfermedad venoclusiva hepática; sin embargo, esta no es apoyada por los hallazgos histopatológicos consignados en el resumen clínico.
Un punto importante es sospechar de hepatitis autoinmune en el paciente, dado que es una causa tratable de hepatopatía crónica. Esto es obligatorio por las características ya descritas además de por la presencia de anticuerpos antimicrosomas, renal-hepático tipo 1 y antinucleares, que incluso en su momento necesitaron tratamiento esteroideo durante 6 meses. Sin embargo, de acuerdo con los criterios diagnósticos establecidos por el grupo internacional de estudio de hepatitis autoinmune, modificados y simplificados en 2008, se debe incluir histología hepática compatible, hipergammaglobulinemia selectiva, positividad de anticuerpos y exclusión de infecciones virales y enfermedad de Wilson. La información con la que se contó no resulta suficiente para integrar o descartar este diagnóstico.
Ante un paciente con los antecedentes ya mencionados, que clínicamente se manifiesta con datos de hipertensión portal, y bioquímicamente con discreta elevación de las aminotrasferasas, mínima colestasis y sin afección en la síntesis hepática, pareciera no concordar el diagnóstico de cirrosis biliar reportado en la biopsia hepática. Sin embargo, existe una entidad que compagina con esta presentación: la fibrosis hepática congénita, en la que las formas más fibróticas son histológicamente confundibles con cirrosis biliar, ya que, habitualmente, suele observarse fibrosis periportal y proliferación irregular de conductos biliares, originando obliteración venosa. La fibrosis hepática congénita frecuentemente se asocia con daño renal, principalmente manifestado por poliquistosis y nefronoptisis. En este paciente no se encontró ninguna afección de este tipo, con excepción de la hematuria. Si bien esta entidad normalmente es de curso benigno, la malformación de la placa ductal causa múltiples grados de hipertensión portal hasta en el 70% de los pacientes afectados, y se convierte en el principal motivo de morbimortalidad de los pacientes1–4.
La edad de presentación habitual es la adolescencia; sin embargo, las series de casos reportadas a nivel mundial muestran una amplia gama de fenotipos, con una herencia autosómica recesiva, y afectación predominantemente hepática, que puede estar presente desde el periodo neonatal hasta la adolescencia y adultez. Teniendo en cuenta lo descrito, es importante enfatizar que el estudio de las hepatopatías crónicas, ante su grado de complejidad, debe realizarse de manera ordenada y sistematizada para lograr un diagnóstico oportuno. Se debe poner especial atención en la presentación clínica ya que, a pesar de ello, en un número significativo de pacientes incluso no se logra definir la etiología de la misma5–8.
La principal complicación que presentó el paciente fue el sangrado de tubo digestivo alto secundario a la hipertensión portal, de tipo intrahepática, debida a la fibrosis periportal y a la compresión sinusoidal. También cursó con progresión en la dilatación venosa prehepática.
Durante los eventos de sangrado del paciente no se mencionó si la intervención endoscópica fue realizada de manera oportuna; esto es, dentro de las primeras 24h de inicio y una vez hecha la reanimación hídrica pertinente. No se realizó ningún tipo de tratamiento en las várices gástricas, ya que por su complejidad representan un reto terapéutico importante porque en estas se pueden emplear adhesivos tisulares. De hecho, tan solo el tratamiento exclusivo en várices esofágicas tiende a agravar las fúndicas previamente existentes, ocasionando mayor riesgo de sangrado.
Con respecto a la transfusión de hemoderivados, contrario al sentido común, no se recomienda la administración de plasma fresco congelado en pacientes con cirrosis y sangrado de tubo digestivo alto con el objetivo de corregir la coagulopatía, ya que esto no ha demostrado beneficio en el pronóstico del paciente. Tampoco está indicado su uso de manera profiláctica, previo a una endoscopia, ya que condiciona el incremento en la presión plasmática, y por tanto portal, aumentando el riesgo de sangrado, como en este caso.
La administración de concentrados plaquetarios está recomendada solamente cuando se tiene un recuento por debajo de 30,000 y una meta de hemoglobina recomendada entre 7-8g/dl.
Ahora bien, durante un sangrado de tubo digestivo bajo presentado como hematoquecia masiva, se recomienda realizar una angiotomografía y un gammagrama, con la finalidad de localizar el sitio del sangrado. De no evidenciarse el lugar, se debe realizar una colonoscopia diagnóstica y terapéutica dado que, hasta en el 75% de los casos, se logra localizar el sitio de sangrado y hasta en el 95% se consigue realizar una hemostasia endoscópica. Por lo anterior, se recomienda ampliamente realizar una colonoscopia durante las primeras 24h de sangrado activo y, después de la intervención endoscópica, utilizar análogos de somatostatina o terlipresina durante 3 días. El paciente no se vio beneficiado por ninguno de estos estudios diagnósticos ni terapéuticos; a pesar de ello, estos tratamientos no se consideran de tipo curativo, sino paliativo.
Respecto de la profilaxis primaria de sangrado de tubo digestivo en pacientes con hipertensión portal y cirrosis biliar, el uso de beta-bloqueadores no selectivos, como el propranolol, tiene poca evidencia demostrada en niños. Sin embargo, en adultos, los estudios han demostrado disminución en la incidencia de sangrado de hasta el 50%, motivo por el cual su uso en pacientes pediátricos es común, como en el presente caso. A pesar de ello, no se ha logrado evidenciar diferencia significativa entre estas dos conductas terapéuticas.
Como profilaxis secundaria, se realizó la ligadura de várices. Con relación a las várices gástricas, el uso de cianoacrilato, un adhesivo tisular, ha mostrado efectividad similar a la ligadura, con una menor tasa de recurrencia de sangrado. Sin embargo, estas son un reto terapéutico a escala mundial. Este tipo de pacientes se consideran candidatos ideales para la realización de cortocircuitos portosistémicos, dado que la función hepática se encuentra conservada. Por esto, se debió plantear la realización de esta derivación vascular, y así disminuir significativamente la morbilidad y, por consiguiente, la mortalidad del paciente. Como en el presente caso no quedaba clara la etiología de la fibrosis, y ante la postura de realizar un trasplante hepático, se considera importante la no manipulación quirúrgica de este sitio. Otra complicación importante que presentó el paciente fue la ascitis, la cual incrementó por la disminución de la presión osmótica y aumento del sistema renina-angiotensina-aldosterona, que condiciona fuga a tercer espacio, principalmente a nivel peritoneal. Este paciente recibió una mezcla de soluciones intravenosas con aportes altos de sodio, lo cual perpetuó e incrementó la ascitis durante una descompensación hemodinámica. El tratamiento de la ascitis pasa a segundo término, y debe ser tomada en cuenta una vez que se ha estabilizado al paciente.
Finalmente, tras 5 años de cursar con eventos de sangrados repetidos, presentó un cuadro infeccioso asociado con encefalopatía hepática, con prolongación de los tiempos de coagulación e INR mayor de 1.5, a pesar de contar con una adecuada administración de vitamina K. Esto se define como una insuficiencia hepática aguda en el contexto de un paciente con hepatopatía crónica.
Dentro del manejo de la encefalopatía hepática, fue correcta la utilización de lactulosa; sin embargo, no se inició hasta la segunda semana de hospitalización. En vez de la lactulosa se pueden emplear antibióticos, como rifaximina, que tienen poca absorción enteral, efectividad similar a esta y menos efectos adversos.
Es importante que, una vez alcanzada la estabilidad hemodinámica, se asegure un adecuado aporte calórico-proteico a fin de evitar el catabolismo. Se recomienda iniciar con una dosis de proteína de 0.5g/kg/día, con aumentos paulatinos hasta alcanzar 1.5g/kg/día. Se debe preferir el uso de proteína vegetal por su menor cantidad de metionina y aminoácidos aromáticos. Todas estas medidas, junto con una adecuada glucemia y equilibro electrolítico, han demostrado ser parte fundamental del tratamiento de la encefalopatía hepática.
No son de extrañar los eventos de infección grave que presentó el paciente, dado que la insuficiencia hepática condiciona una menor producción de inmunoglobulinas y factores del sistema del complemento, afectando con ello la inmunidad innata y adaptativa del paciente. De igual forma, el hiperesplenismo condiciona la disminución en la cuenta y función leucocitaria, llevando a un estado de inmunocompromiso secundario agravado por los múltiples antecedentes previamente mencionados.
Un punto fundamental sobre este paciente es mencionar el beneficio que hubiese presentado de haberse realizado el trasplante hepático. Se ha descrito que hasta el 7% de los pacientes con fibrosis hepática congénita que superan el periodo neonatal requerirán de un trasplante para resolver las complicaciones por hipertensión portal y colangitis recurrentes. Dos puntos importantes para reflexionar son el largo periodo trascurrido entre el primer contacto con el instituto y el inicio del protocolo de trasplante: si bien fue conocido por la mayoría de los servicios involucrados, no se logró completar durante los 35meses posteriores al inicio. El paciente mostró deterioro hemodinámico y ventilatorio, tanto por los procesos infecciosos como por los múltiples sangrados, progresando a una falla multiorgánica (tabla 1).
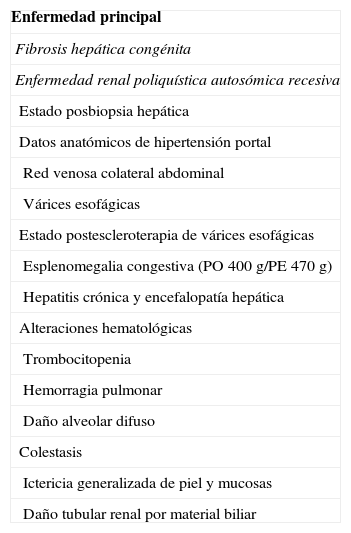
Diagnósticos anatómicos finales
| Enfermedad principal |
| Fibrosis hepática congénita |
| Enfermedad renal poliquística autosómica recesiva |
| Estado posbiopsia hepática |
| Datos anatómicos de hipertensión portal |
| Red venosa colateral abdominal |
| Várices esofágicas |
| Estado postescleroterapia de várices esofágicas |
| Esplenomegalia congestiva (PO 400g/PE 470g) |
| Hepatitis crónica y encefalopatía hepática |
| Alteraciones hematológicas |
| Trombocitopenia |
| Hemorragia pulmonar |
| Daño alveolar difuso |
| Colestasis |
| Ictericia generalizada de piel y mucosas |
| Daño tubular renal por material biliar |
Los diagnósticos finales son los siguientes:
- •
Cirrosis biliar, probablemente secundaria a fibrosis hepática congénita, lo que condicionó hipertensión portal, várices esofágicas, fúndicas y gastropatía hipertensiva
- •
Sangrado de tubo digestivo alto y bajo
- •
Insuficiencia hepática aguda, en el contexto de hepatopatía crónica
- •
Talla baja, con relación peso/edad y peso/talla no valorable por organomegalias y ascitis
- •
Choque séptico con foco intestinal y pulmonar
Causa inmediata de la muerte:
- •
Hipoxia por insuficiencia respiratoria secundaria a sangrado pulmonar masivo
De los estudios de imagen que se encontraron se mencionan los cambios más importantes. En la imagen del 21 abril de 2010 hay presencia de una cánula en el interior del estómago; en el tórax se observó un leve aumento de la radioopacidad intersticial bilateral, y leve ensanchamiento del mediastino. TAC del sistema nervioso central, sin hallazgos relevantes. telestudios de rayos X de tórax del 23 de abril muestran aumento de la radioopacidad parahiliar bilateral, más aparente en lóbulo superior izquierdo. La proyección toracoabdominal del 21 de octubre muestra distensión del abdomen, alteración en la distribución del gas, predominantemente hacia la zona central, y también aumento del volumen del hígado, que rebasa el reborde costal y está próximo a la parrilla costal del lado izquierdo. El 10 de enero de 2011 se encontró aumento de la radioopacidad parahiliar en el pulmón derecho, sin llegar a la periferia. El 23 de abril de 2011 la imagen del tórax muestra aumento de la radioopacidad del pulmón izquierdo, asimetría de hemidiafragmas y ensanchamiento de mediastino en el perfil izquierdo.
3Discusión (Dr. Alejandro Hernández Plata)Como se ha expuesto anteriormente, dada la complejidad de este caso no fue fácil llegar a un diagnóstico etiológico de la afección hepática. Por ello, la cuestión sería conocer si había elementos suficientes para sospechar hepatitis autoinmune. Actualmente, si se sospecha el diagnóstico de hepatitis autoinmune, la pregunta sería ¿es válido darle tratamiento?
3.1Gastroenterología (Dr. Rodrigo Vázquez)En función de los datos de la historia clínica y los de la biopsia hepática, el diagnóstico de hepatitis autoinmune era poco probable. El conocimiento acerca de cómo diagnosticar y tratar la hepatitis autoinmune ha ido en aumento, pero al ser una causa de hepatopatía crónica se debe tomar en cuenta, ya que en la hepatitis autoinmune es factible dar tratamiento y ver si hay una mejoría clínica. Aunque no es un criterio, podría apoyar el diagnóstico. Con fundamento en la historia clínica, el daño hepático correspondió a cirrosis hepática, pero no se demostró actividad inflamatoria. Se descartaría la presencia de anticuerpos D, ya que en un hígado dañado puede haber elevación de los mismos. Según esto, no sería una hepatitis autoinmune.
Si se tienen criterios clínicos, de laboratorio e histológicos que sugieran el diagnóstico se puede iniciar el tratamiento y valorar la respuesta.
3.2Trasplantes (Dr. Alejandro Hernández Plata)Parte del tratamiento de la hepatopatía crónica es tratar las complicaciones de la hipertensión portal, como son las várices esofágicas. Surgen las siguientes preguntas: ¿cuándo hacer una endoscopia?; ¿cómo tratar las várices esofágicas? y ¿cuál es su seguimiento?
3.3Cirugía de tórax y endoscopia (Dr. Iván Rivas Rivera)Primero se debe definir lo que es la profilaxis primaria y la profilaxis secundaria. Profilaxis primaria son todas aquellas medidas farmacológicas y endoscópicas que se realizan en el paciente que ya tiene confirmada la presencia de várices esofágicas. El uso de beta-bloqueadores no selectivos en pacientes pediátricos está aún en discusión, a diferencia de los pacientes adultos, en quienes está completamente probado. Como sucede a menudo, en población pediátrica faltan estudios aleatorizados y controlados para recomendar los tratamientos. En relación con la endoscopia, se ha llegado a la conclusión de que la esclerosis de las várices en un paciente que no ha sangrado no es un tratamiento adecuado. No así la ligadura de las várices. Este procedimiento tiene la limitación del equipo que se utiliza. Para ligar una várice se necesita un barril (elemento mecánico necesario para el tratamiento de várices esofágicas) especial, que por el tamaño hace que se limite el uso a pacientes de más de 13 a 15kg. Si es posible pasar el endoscópico con el barril, es factible hacer una ligadura de várices de manera profiláctica. La profilaxis secundaria se practica en el paciente que tiene várices pero ya sangró. Se debe seguir con tratamiento farmacológico y a base de esclerosis. Es muy importante descartar la presencia de várices gástricas antes de dar tratamiento a las várices esofágicas. Si hay várices gástricas grandes, con riesgo de sangrado, y se da tratamiento a las várices esofágicas, se corre el riesgo de aumentar el flujo a las várices gástricas y que estas sangren. El tratamiento con cianoacrilato en pacientes pediátricos no está completamente validado. Solamente se recomienda en ciertos pacientes; por ejemplo, en aquellos que no tienen várices esofágicas, o con várices gástricas. En conclusión, hay que tener muy claro el tipo de profilaxis que se le puede aplicar al paciente, para ver si es candidato a esclerosis o si es candidato a ligadura de várices.
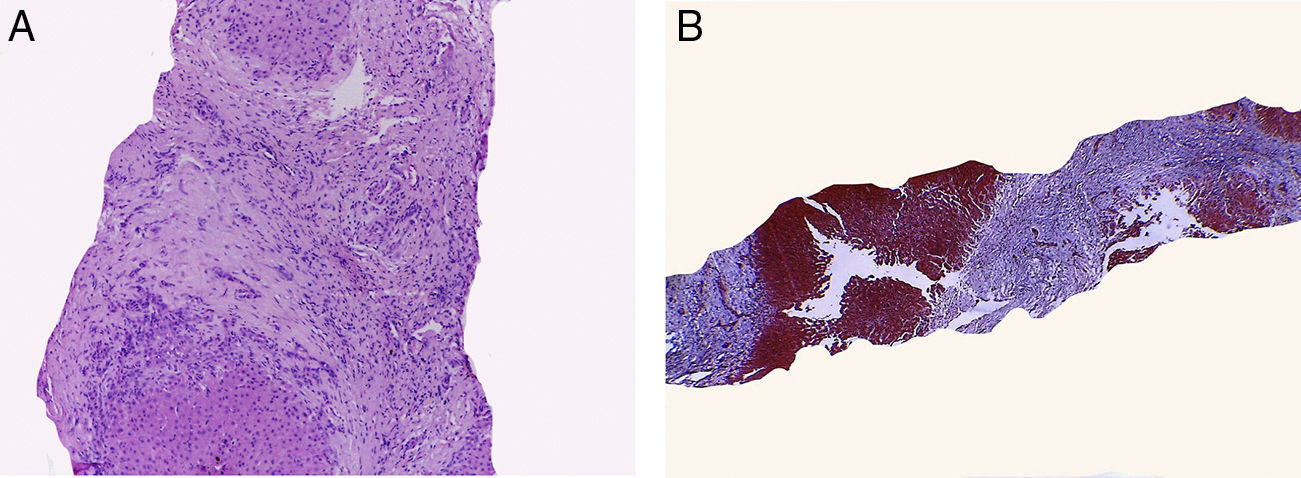
4Patología (Dr. Mario Perezpeña Diazconti)Como ya se mencionó en la historia clínica, el primer contacto que se tuvo con el paciente fue una biopsia hepática percutánea. Se obtuvo un cilindro en el cual pudimos observar que la mayor parte de la biopsia correspondió a tejido fibroconectivo y pequeños nódulos de hepatocitos (fig. 1A). Con la tinción tricrómico de Masson se observó en azul todo lo que corresponde a fibrosis y en rojo los pequeños remanente de hepatocitos.
 La biopsia hepática muestra extensa fibrosis con pequeños nódulos remanentes de hepatocitos, los cuales no presentan alteración. B) El diagnóstico fue de cirrosis biliar secundaria a obstrucción parcial del flujo biliar.")
La biopsia hepática tiene mejor resultado cuando se trata de enfermedades hepáticas difusas, ya que se pueden observar los cambios de la enfermedad en cualquier parte de la biopsia. Sin embargo, en particular en la enfermedad que presentó el paciente, hay más daño en la región subcapsular y en el lóbulo izquierdo del hígado. Por otra parte, hay ocasiones en las que es muy difícil llegar a un diagnóstico ya que el hígado reacciona de la misma manera ante diferentes estímulos. En este caso se realizó el diagnóstico de cirrosis biliar secundaria a obstrucción parcial del flujo biliar (fig. 1B). No se debe pensar inmediatamente en atresia de vías biliares, ya que hay una gran cantidad de enfermedades que pueden dar este patrón. Con la microscopia electrónica se descartaron algunas enfermedades metabólicas y mitocondriales. El reporte de microscopia electrónica fue de cambios colestásicos y de hipertensión portal. Al momento de hacer el estudio post mórtem, se halló líquido hemático en las dos cavidades pleurales. Se hizo una exploración completa en busca de alguna obstrucción, la cual fue descartada.
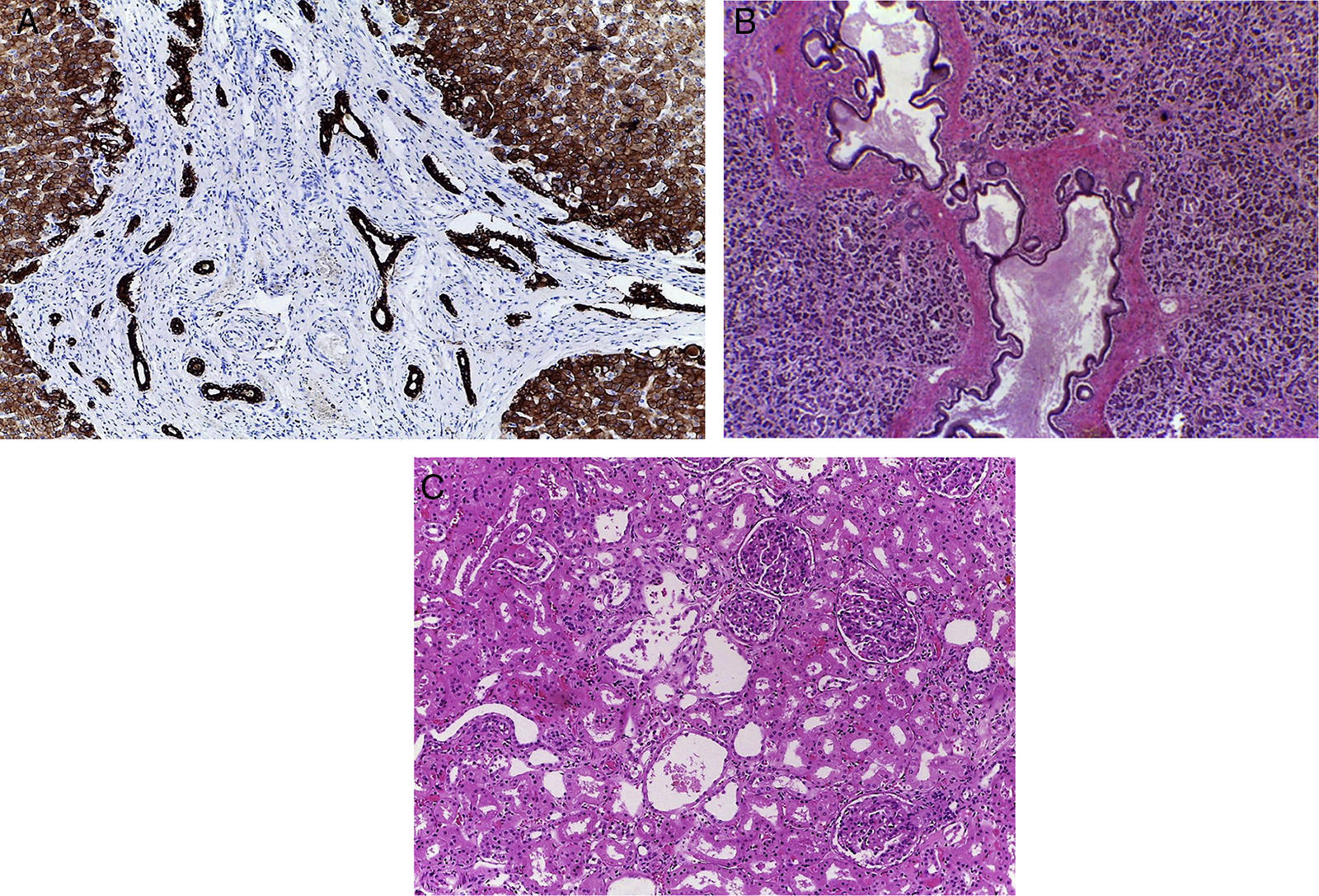
El parénquima hepático mostró una gran cantidad de septos de aspecto fibroso de color amarillo blanquecino; el grosor fue mayor por debajo de la cápsula de Glisson, formó pequeños nódulos y esto explica por qué se vio la biopsia con gran cantidad de fibrosis (fig. 2). Esta enfermedad afecta con mayor intensidad la periferia del órgano, y disminuye hacia la parte central del hígado. Como ya se mencionó, es un caso de fibrosis hepática congénita. Esta enfermedad está causada por una alteración en una proteína llamada fibrocistina. La alteración citogenética es una mutación en el brazo corto del cromosoma 6, donde está el gen PKHD1 (de enfermedad poliquística renal y hepática). La disfunción de esta proteína afecta los conductos biliares del hígado, los conductos colectores del riñón y los conductos pancreáticos (fig. 3). Histológicamente, hay fibrosis en los espacios porta y formación de conductos irregulares, quísticos, por una fusión de la placa ductal. La placa ductal son dos capas de células que se disponen alrededor de una vena porta durante su fusión y remodelación. Cuando hay una falla de esta proteína aumenta el número de conductos biliares que se forman. Los conductos biliares intrahepáticos, los cuales están dilatados, son tortuosos, y el epitelio está aplanado. Las venas porta también muestran alteración: están aumentadas en número y son pequeñas.

Se observa el hígado, la vesícula biliar y las vías biliares extrahepáticas que fueron exploradas y se encontraron permeables. Además, se muestra el páncreas y duodeno. En el parénquima hepático hay abundantes septos delgados de tejido fibroconectivo, que conforme se acercan a la periferia son de mayor grosor. La cápsula es fibrosa.
 los conductos hepáticos, B) pancreáticos y C) túbulos renales.")
En el páncreas, los conductos están dilatados y con material eosinófilo denso en su luz. Se observa, además, necrosis del componente epitelial y del tejido adiposo.
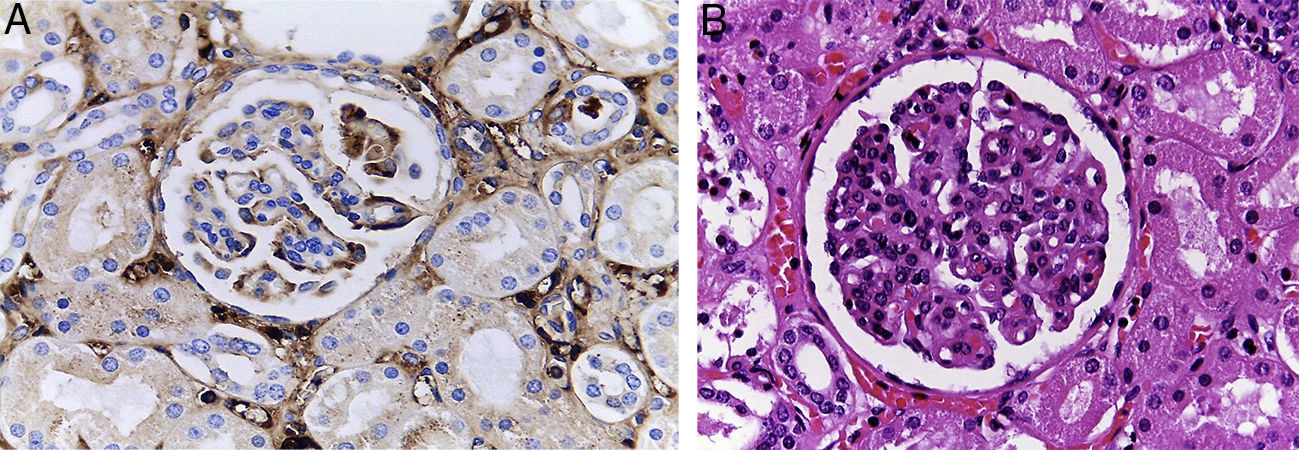
El otro órgano afectado es el riñón (fig. 4). Ambos riñones están aumentados de tamaño y peso, como se ha descrito en la enfermedad renal poliquística autosómica recesiva. Histológicamente, se observan pequeños quistes originados en los túbulos colectores revestidos por epitelio cúbico aplanado, y corresponden a las ramas terminales de los túbulos colectores. En esta enfermedad, cuando los cambios renales son más aparentes, el daño hepático es menor y viceversa.

Debido al daño hepático, el riñón sufre otro cambio. Es una glomerulonefritis focal, proliferación mesangial por depósitos de IgA, cirrosis hepática e insuficiencia hepática, y daño por material biliar en lo túbulos del riñón.
También por el daño hepático, en el esófago y estómago se encuentran várices dilatadas y congestivas asociadas con zonas ulceradas en el esófago. En estas zonas se halla fibrina, detritus celular y levaduras, que pueden corresponder a Candida sp. En el estómago se encontraron 35ml de sangre.
Los pulmones se encontraron aumentados de tamaño y peso, con extensas zonas de color café rojizo. Los cortes histológicos mostraron extensas zonas de hemorragia pulmonar extensa. No se encontraron datos de infección. Focalmente hay membranas hialinas asociadas con daño alveolar. En la pleura se observó edema y vasos linfáticos dilatados. El bazo estaba aumentado de tamaño y peso con congestión pasiva crónica.
En conclusión, se trató de un paciente con fibrosis hepática congénita en la cual el daño renal y pancreático estuvo presente de manera discreta.
5Comentarios finales5.1Nefrología (Dr. Luis Velásquez Jones)La mayoría de los estudios en pacientes con hepatopatía crónica que desarrollan una afección renal se han hecho en adultos. Se han realizado muy pocos estudios en población pediátrica. En adultos, el 50% de los pacientes con cirrosis tiene elevación de IgA. En este paciente se observó la elevación de IgA. Se sabe que los pacientes adultos cirróticos desarrollan proliferación mesangial en el riñón; además, en la inmunofluorescencia se encuentran depósitos abundantes de IgA, que correlacionan con los niveles séricos de IgA elevados, observados en el 90% de estos pacientes. En este caso, el paciente presentó hematuria y niveles elevados de IgA. Se puede decir con bastante certeza que tuvo una nefropatía por IgA con elevación de la creatinina. Esta enfermedad no es frecuente, aunque se ha informado nefropatía IgA secundaria en pacientes con atresia de vías biliares. Cuando se hace el trasplante renal en estos pacientes, la nefropatía por IgA desaparece.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.