




Se presenta el caso de una niña de 7 meses que cursa con falla para crecer y alteraciones metabólicas inespecíficas.
1.1AntecedentesMadre de 21 años, padre de 24 años, sanos, sin datos de consanguinidad. Hermano de 3 años, sano. Alimentación al seno materno durante un mes, y alimentación complementaria desde los cinco meses.
Desarrollo Psicomotor. Sostén cefálico parcial, no gira en su posición, sin sedestación, acude a terapia de rehabilitación.
Inmunizaciones. BCG, una dosis de pentavalente acelular y dos dosis de hepatitis B.
Producto G II, control prenatal desde el primer mes, con ingesta de hierro y ácido fólico. Parto hospitalario a las 40 SDG, pesó 3,050g, midió 49cm, Apgar 8/9. Egresó a las 48 horas sin la madre, quien permaneció hospitalizada cinco días por choque hipovolémico.
La paciente fue hospitalizada a los 2 meses de vida durante 15 días por gastroenteritis aguda con deshidratación; recibió antibióticos y una transfusión de glóbulos rojos por anemia. A los 5 meses de edad fue hospitalizada por presentar aumento de perímetro abdominal. Se detectó hepatomegalia por ultrasonido, por lo que se refirió al Hospital Infantil de México Federico Gómez (HIMFG). Se registraron peso de 4,800g, talla de 57cm y perímetro cefálico de 41cm. Se reportó borde hepático a 4-3-4cm debajo de reborde costal.
Al ingreso al HIMFG se registró aumento de perímetro abdominal de 46 a 50cm de 8 días de evolución, con incremento de red venosa colateral, e ictericia de 4 días previos, progresiva, en mucosas y escleras.
Se observó deterioro neurológico progresivo de 2 días con hipoactividad, somnolencia y estupor, con aumento en horas de sueño, sin respuesta al despertarla. Se registraron los siguientes datos: peso 4,000g, talla 61cm, frecuencia cardiaca 153/min, frecuencia respiratoria 30/min, presión arterial 80/50mmHg, temperatura 37.7°C, perímetro abdominal 53cm, calificación de la escala de Glasgow 9.
Paciente de sexo femenino de edad aparente menor a la cronológica, somnolienta. Normocéfala, pupilas isocóricas, normorrefléxicas, escleras ictéricas. Cuello sin adenomegalias. Tórax con asimetría en movimientos de amplexión y amplexación, campos pulmonares con adecuada entrada y salida de aire. Ruidos cardiacos aumentados en frecuencia, sin soplos. Abdomen globoso a expensas de ascitis, blando, depresible, no doloroso, con peristalsis. Extremidades íntegras, buen llenado capilar, pulsos adecuados en intensidad. Apertura ocular, respuesta verbal y motora solo ante estímulo doloroso.
En una revisión oftalmológica se describió catarata nuclear en ambos ojos.
La paciente permaneció hospitalizada en estado crítico durante dos meses. A su ingreso se aseguró la vía aérea con cánula endotraqueal, y aunque pudo ser extubada, presentó deterioro ventilatorio por la restricción asociada con la ascitis, la mala distribución de líquidos y por eventos infecciosos nosocomiales. La estrategia ventilatoria fue dinámica e incluyó ventilación de alta frecuencia al final de su estancia.
Presentó varios eventos de inestabilidad hemodinámica, algunos relacionados con datos de infección nosocomial corroborada. Requirió manejo con vasopresores e inotrópicos. En uno de los hemocultivos se aisló Enterococcus faecium resistente a ampicilina. Se manejó con diversos esquemas antibióticos de amplio espectro a dosis meníngeas por la imposibilidad de descartar infección de sistema nervioso central. Ante los factores de riesgo de la paciente y la aparición de hifas en orina, también se incluyó cobertura antifúngica.
Desde el punto de vista metabólico, la paciente cursó con hiponatremia de difícil manejo e hipoglucemia. En lo hematológico, ingresó con anemia y se reportó anisocitosis, macrocitosis y acantocitosis en el frotis de sangre periférica. Tuvo sangrado de mucosas por plaquetopenia y coagulopatía secundarias a falla hepática y a coagulación intravascular diseminada, que se manejó con vitamina K, trasfusión de plasma y de crioprecipitados (tabla 1).
Biometría hemática, tiempos de coagulación y química sanguínea
| Hb | Hto | Leu | Seg | Ban | Linf | Mon | Alfa 1 antitripsina |
|---|---|---|---|---|---|---|---|
| 7.10 g/dl | 22.8% | 37,300 | 42% | 8% | 24% | 26% | 106 mg/dl |
| TP | TTP | INR | DHL | Colesterol | Triglicéridos | HDL | LDL |
|---|---|---|---|---|---|---|---|
| 31.6” | >120” | 2.43 | 477 U | 67 mg/dl | 38 mg/dl | 33 mg/dl | 26.4 mg/dl |
| BUN | Creat | BD | BI | BT | ALT | AST | DHL | Alb | Prot T |
|---|---|---|---|---|---|---|---|---|---|
| 19 mg/dl | 0.5 mg/dl | 10.6 mg/dl | 12.7 mg/dl | 23.3 mg/dl | 27 U | 40 U | 524 U | 4 g/dl | 5.2 g/dl |
| BD | BI | BT | ALT | AST | Alb | Prot T | Gluc | Amonio | GGT |
|---|---|---|---|---|---|---|---|---|---|
| 9.9 mg/dl | 12.7 mg/dl | 22.6 mg/dl | 31 U | 75 U | 3 g/dl | 8.3 g/dl | 61 mg/dl | 144-259μmol/l | 27 U |
Hb: hemoglobina; Hto: hematocrito; Leu: leucocitos; Seg: segmentados; Ban: bandas; Linf: linfocitos; Mon: monocitos; TP: tiempo de protrombina; TTP: tiempo parcial de tromboplastina; INR: relación internacional normalizada; DHL: deshidrogenasa láctica; HDL: lipoproteína de alta densidad; LDL: lipoproteína de baja densidad; BUN: nitrógeno ureico en sangre; Creat: creatinina; BD: bilirrubina directa; BI: bilirrubina indirecta; BT: bilirrubina total; ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; DHL: deshidrogenasa láctica; Alb: albúmina; Prot T: proteína total; Gluc: glucosa; GGT: gamma glutamil transferasa.
En cuanto al abordaje y manejo de la hepatopatía, se identificó hipertensión portal intrahepática sin esplenomegalia. Evolucionó con incremento del perímetro abdominal, elevación de aminotransferasas y amonio, hipoglucemia, sin acolia ni coluria (tabla 1). Se descartaron causas estructurales e infecciosas de la hepatopatía (tabla 2). Se reportó perfil de aminoácidos y acilcarnitinas normal. Los niveles de tirotropina (TSH) fueron de 0.9μUI/ml y los de 25-OH vitamina D, de 27.7 ng/ml.
Estudios de serología
| VHA | VHB | VHC | VIH | Toxoplasma | Rubeola | ||
|---|---|---|---|---|---|---|---|
| IgM | IgG | Ag Sup | Ac Antígeno Sup | - | - | IgG/IgG | IgG/IgM |
| - | + | - | + | - | <10 U/ml | ||
| CMV | VHS I | VHS II | Ag. EB | ||||
| IgG | IgM | IgG | IgM | IgG | IgM | IgG | EBNA |
| - | - | + | - | + | <10 U/ml | <10 U/ml | <5.0 U/ml |
| IgA total | IgE total | IgM total | IgG total | ||||
| 10.9 g/dl | <17.3 UI/ml | 260 mg/dl | 1,120 mg/dl | ||||
VHA: virus de hepatitis A; VHB: virus de hepatitis B; VHC: virus de hepatitis C; VIH: virus de la inmunodeficiencia humana; CMV: citomegalovirus; VHS I: virus de herpes simple I; VHS II: virus de herpes simple II, EB: virus de Epstein-Barr.
En los periodos sin sedación, la escala de Glasgow era de 12. Se indicaron restricción de sodio a 2 mEq/kg/d, espironolactona 6mg/kg/d, furosemide 1mg/kg/d, lactulosa 1ml/kg/d, rifaximina 20mg/kg/d, vitamina K 10mg/d, plasma fresco congelado y crioprecipitados. Se restringió la ingesta de proteínas. Por persistencia de ascitis y patrón ventilatorio restrictivo se inició infusión de albúmina a 1g/kg.
La función renal se deterioró. La creatinina sérica se elevó hasta 0.7mg/dl (tabla 3), con fracción excretada de sodio (FeNa) de 0.6 y el sodio urinario de 6 mmol/l. Se diagnosticó síndrome hepatorrenal que se manejó con terlipresina e infusión de albúmina. La presión intrabdominal se registró en 13cm/H2O; se realizó paracentesis con drenaje de 1,200ml, con lo que mejoraron los azoados y aumentó el volumen urinario.
Pruebas de función renal
| BUN | Creat | Na | K | Cl | Ca | P | Mg | |
|---|---|---|---|---|---|---|---|---|
| Sangre | 7.5 mg/dl | 0.5 mg/dl | 146 mEq/l | 4.1 mEq/l | 101 mEq/l | 8.3 mg/dl | 3.7 mg/dl | 1.9 mg/dl |
| Orina | 204 mg/dl | 10.2 mg/dl | 50 mEq/l | 20.9 mEq/l | 54 mEq/l | 9.3 mg/dl | 36.3 mg/dl | |
| Examen general de orina | Cetonas + | Glucosa + | ||||||
BUN: nitrógeno ureico en sangre; Creat: creatinina; Na: sodio; K: potasio; Cl: cloruro; Ca: calcio; P: fosfatos; Mg: magnesio.
El servicio de algología indicó morfina a 100μg/kg/dosis. Después de dos meses de estancia intrahospitalaria con manejo intensivo presentó paro cardiorrespiratorio. No se realizaron maniobras de reanimación por solicitud de los padres.
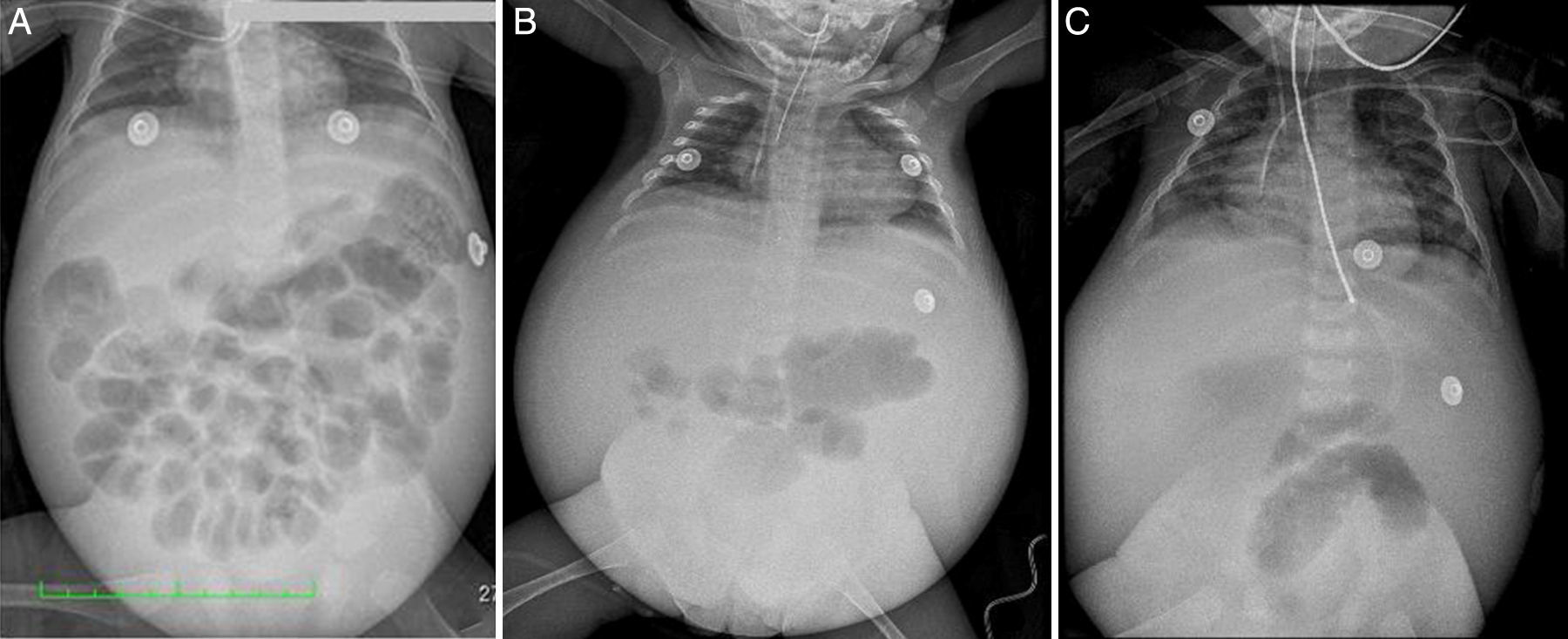
2ImagenologíaEn las placas toracoabdominales tomadas durante su evolución, se observó desplazamiento de asas de intestino en sentido cefálico, radiopacidad en hueco pélvico, dilatación de asas de intestino y volumen pulmonar disminuido por elevación del diafragma. La radiopacidad difusa en abdomen incrementó con el tiempo en relación con líquido libre de ascitis. Se observó horizontalización de los arcos costales y el corazón mostró crecimiento de cavidades (fig. 1).

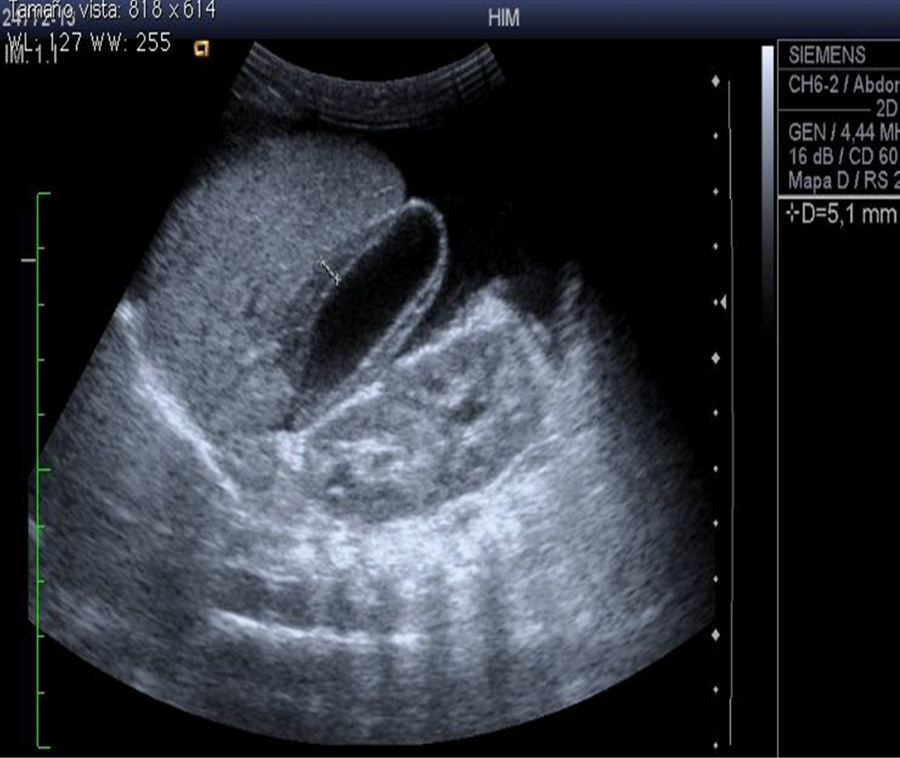
En el ultrasonido abdominal se observó el hígado con ecogenicidad heterogénea y bordes microlobulados, y la vena porta de 4.8mm. Flujo hepatópeta de 26cm/s, colaterales en hilio con flujo helicoidal. Arteria hepática con IR 0.9 y venas suprahepáticas permeables. Se concluyó daño hepático crónico, hipertensión portal y probable degeneración cavernomatosa de la misma (fig. 2).

En la tomografía axial computarizada (TAC) de cráneo simple se observó un aumento de la profundidad de los surcos y cisuras con incremento del volumen compensatorio del sistema ventricular y quiste aracnoideo. Sin evidencia de hemorragia ni isquemia.
3Presentación del casoLos diagnósticos sindromáticos se presentan a continuación.
- •
Síndrome de hipertensión portal: de tipo intrahepático, con base en ascitis, red venosa colateral, hallazgos ultrasonográficos con reporte de venas suprahepáticas y porta permeables.
- •
Síndrome colestásico: con base en ictericia y valores séricos de bilirrubina directa mayores a 2mg/dl.
- •
Síndrome de respuesta inflamatoria sistémica: con base en fiebre, taquicardia y leucocitosis.
- •
Síndrome encefálico: con base en la alteración del ciclo sueño/vigilia, irritabilidad y disminución del estado de alerta, sin datos de focalización.
- •
Síndrome hemorragíparo: con base en sangrado en sitios de punción, gingivorragia, sangrado por cánula endotraqueal, gasto hemático por sonda orogástrica, hemoglobina y hematocrito por debajo de valores normales para la edad, trombocitopenia y tiempos de coagulación prolongados.
- •
Síndrome de dificultad respiratoria: con base en tiraje intercostal, aleteo nasal y disociación toracoabdominal.
- •
Síndrome compartimental abdominal: con base en incremento del perímetro abdominal y presión intraabdominal con deterioro de la función renal.
Los diagnósticos nosológicos fueron los siguientes:
- •
Encefalopatía hepática con base en el deterioro neurológico e hiperamonemia.
- •
Insuficiencia hepática con base en INR > 1.5, sin mejoría tras la administración de vitamina K, hipoglucemia, hipoalbuminemia y encefalopatía hepática.
- •
Insuficiencia de vitamina D por concentración de vitamina D entre 21-29 ng/dl.
- •
Sepsis con base en datos de síndrome de respuesta inflamatoria, manifestaciones abdominales y resultado de cultivos.
- •
Tubulopatía proximal con base en glucosuria, hipofosfatemia, hipokalemia e hiperfosfaturia.
- •
Insuficiencia renal prerrenal con base a elevación de creatinina sérica, urea y FeNa de 0.6.
- •
Coagulación intravascular diseminada con base en síndrome hemorragíparo, trombocitopenia, prolongación de tiempos de coagulación y disminución de fibrinógeno.
- •
Choque séptico con base en datos de respuesta inflamatoria sistémica, hipotensión, extremidades frías, pulsos débiles y necesidad de infusión de norepinefrina.
- •
Falla orgánica múltiple por disfunción cardiovascular, respiratoria, hematológica, hepática, renal y neurológica.
En cuanto a la etiología de las manifestaciones hepáticas, las causas estructurales de hipertensión portal eran poco probables ya que no contaba con antecedentes de acolia, hipocolia o coluria. Se descartaron también las causas infecciosas más comunes de hepatopatía, pues la serología fue negativa para virus de hepatitis A, B, C, citomegalovirus (CMG), virus de Epstein-Barr, toxoplasma, rubeola y VIH. Algunas etiologías menos frecuentes que no se descartaron fueron infección por Echovirus, virus de Coxsackie, adenovirus, parvovirus y varicela. También se descartó hipotiroidismo.
La hepatopatía de rápida progresión a insuficiencia hepática, el retraso en el desarrollo psicomotor, la tubulopatía y la catarata nuclear bilateral hicieron sospechar de una etiología metabólica. Los defectos de la beta-oxidación se descartaron por la presencia de cetonuria y nitrógeno ureico normal. No era probable un trastorno en el metabolismo de los hidratos de carbono puesto que, mientras se encontró hemodinámicamente estable, no tuvo datos de hipoglucemia, acidosis metabólica ni hiperlactatemia. No se contó con la determinación de galactosa total, sustancias reductoras en orina ni biopsia hepática, para eliminar este grupo de enfermedades.
La alcalosis respiratoria, cetonuria y el amonio mayor a 100μmol/l hacen pensar en un defecto en el ciclo de la urea, pero sería necesaria la determinación específica del déficit enzimático para su confirmación. Por otro lado, aunque se describió un perfil de aminoácidos normal, no se contó con succinil acetona y ácidos orgánicos en orina para eliminar la probabilidad de una aminoacidopatía.
Por lo comentado anteriormente, las posibilidades diagnósticas eran galactosemia, glucogenosis, tirosinemia o un defecto en el ciclo de la urea. Además, considerando los niveles normales de gamma glutamil transpeptidasa, otras alternativas eran una alteración en la síntesis de ácidos biliares o colestasis intrahepática familiar progresiva.
Vale la pena comentar que esta paciente también presentaba falla en el crecimiento (talla para la edad al nacimiento del 98% con disminución al 89% a los 5 meses de edad), y además padecía desnutrición aguda grave de acuerdo con los criterios de la OMS para pacientes de 6 a 60 meses (peso para la talla menor al 70%).
En esta paciente debió sospecharse peritonitis bacteriana espontánea a su ingreso a urgencias debido al aumento del perímetro abdominal de 7cm con respecto a la primera valoración, ictericia progresiva con aumento en bilirrubina directa y prolongación de los tiempos de coagulación. Para corroborar este diagnóstico estaba indicada la paracentesis y el envío de líquido peritoneal a cultivo.
La hiponatremia en pacientes hepatópatas con ascitis es generalmente de tipo dilucional, por retención renal de sodio y agua. Las principales indicaciones para iniciar el manejo para la hiponatremia son síntomas neurológicos o sodio sérico inferior a 120 mEq/l. La restricción de líquidos es la opción principal para lograr un aumento sostenido en el sodio sérico. Además, si se presenta hipokalemia, como en este caso, debe corregirse ya que el sodio sérico tenderá a elevarse.
La ascitis refractaria se define como aquella que no puede ser movilizada o que presenta recurrencia temprana. Ocurre del 5-10% de los pacientes y se subdivide en dos grupos: resistente a diuréticos e intratable a diuréticos. La paciente se encontraba en este último grupo, ya que durante su evolución cursó con insuficiencia renal que contraindicaba el manejo con altas dosis de diuréticos.
Durante su evolución, la paciente cursó con elevación de la creatinina, sin mejoría después de la expansión de volumen y uso de albúmina. Ante un paciente con cirrosis más ascitis en ausencia de enfermedad parenquimatosa renal y sodio urinario en 6 mmol/l, se debe sospechar síndrome hepatorrenal. Sin embargo, la paciente no cumplía con los criterios establecidos para su diagnóstico, ya que existía el antecedente de uso de medicamentos nefrotóxicos, diuréticos y la creatinina sérica no alcanzaba un valor de 1.5mg/dl.
Los diagnósticos finales fueron los siguientes:
- •
Hepatopatía, probablemente secundaria a error innato del metabolismo
- •
Hipertensión portal intrahepática
- •
Insuficiencia hepática aguda
- •
Encefalopatía hepática grado III
- •
Ascitis intratable
- •
Sepsis con foco abdominal
- •
Desnutrición aguda grave
- •
Retardo en el crecimiento de intensidad moderada
- •
Retraso en el desarrollo psicomotor
Se realizó la autopsia de una lactante menor con ictericia generalizada, datos de hipertensión portal caracterizados por distención abdominal secundaria a ascitis, red venosa colateral toracoabdominal, protrusión del ombligo y disminución generalizada de la masa muscular. A la apertura de cavidades se encontraron 950ml de líquido cetrino peritoneal y hemorragias epicárdicas.
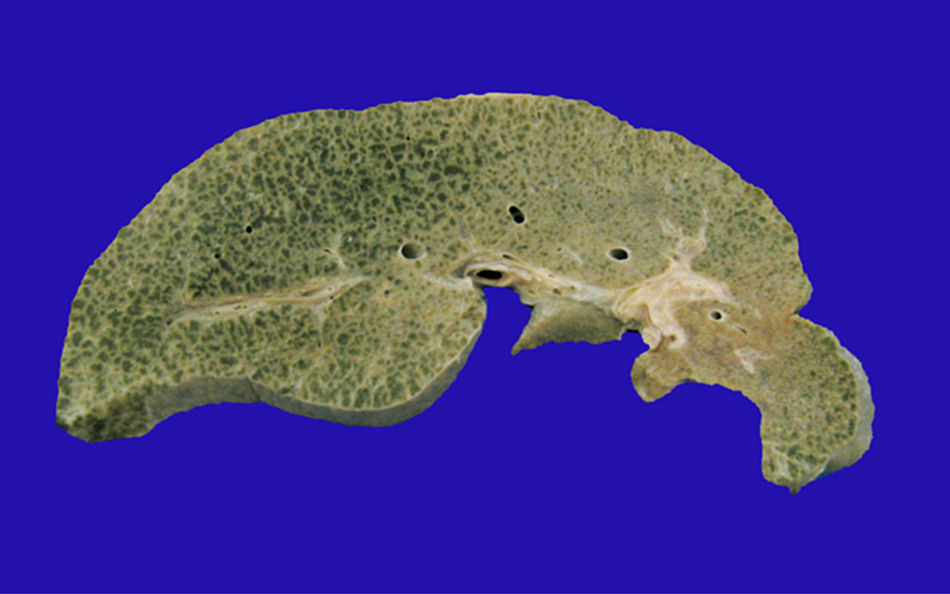
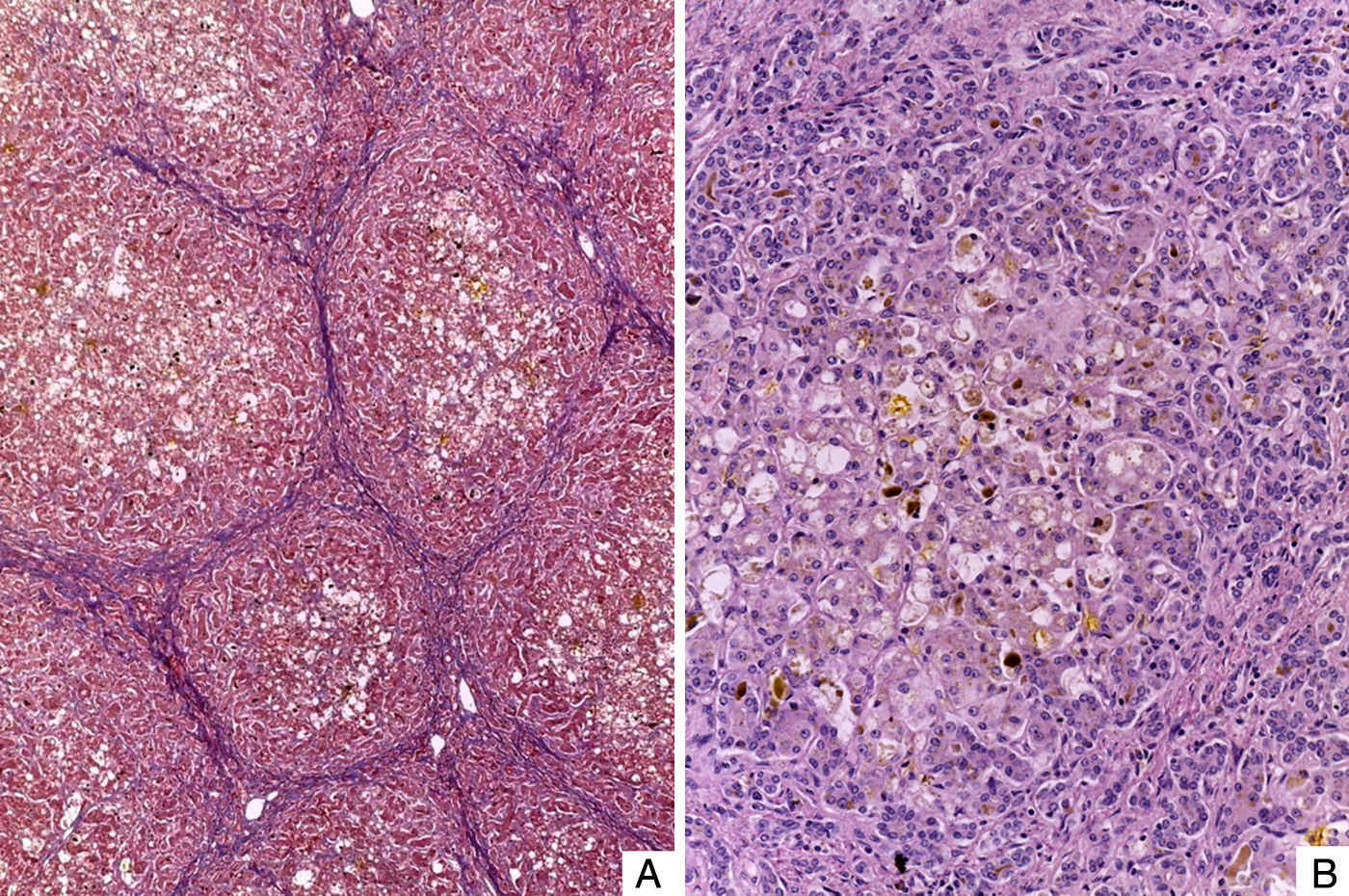
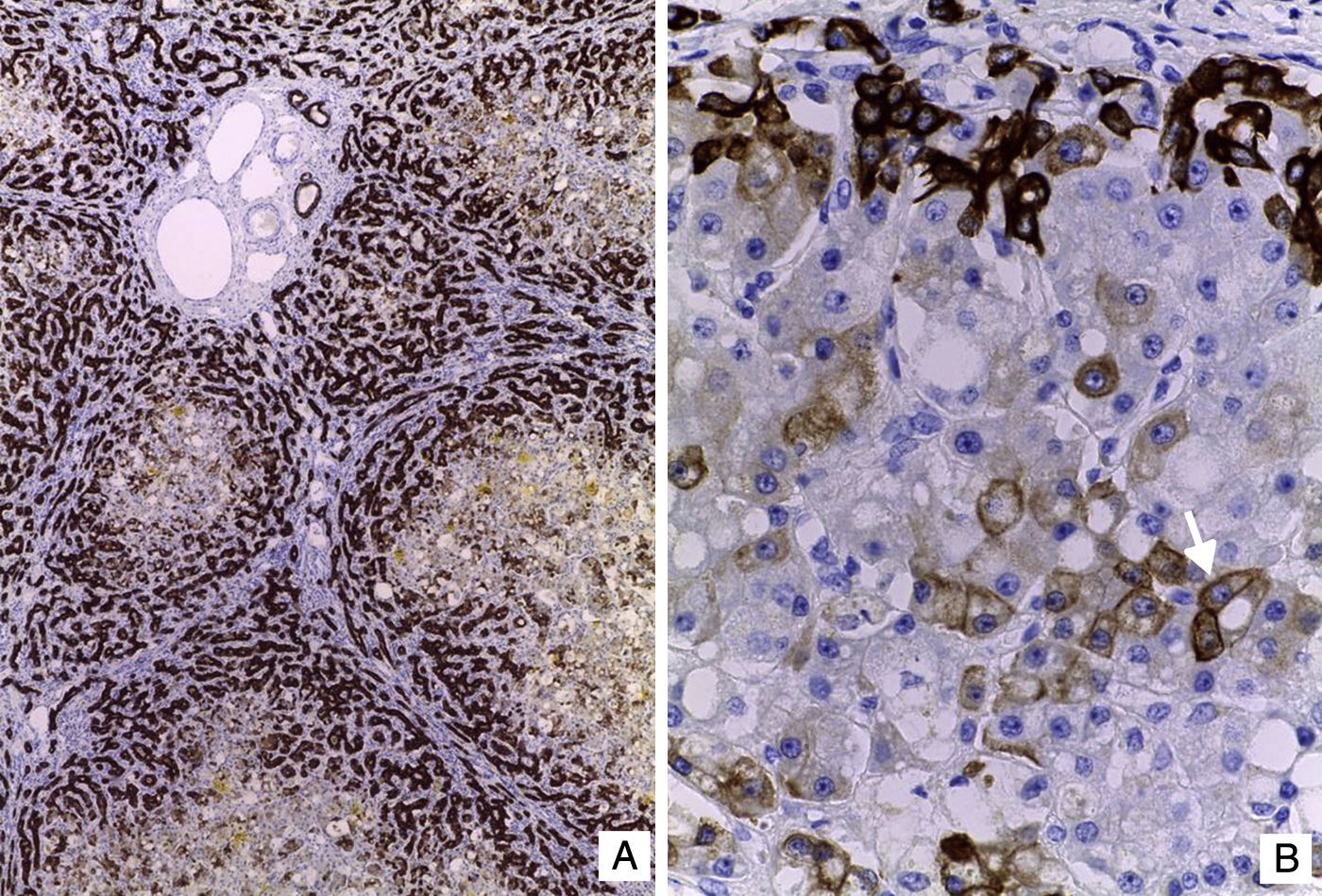
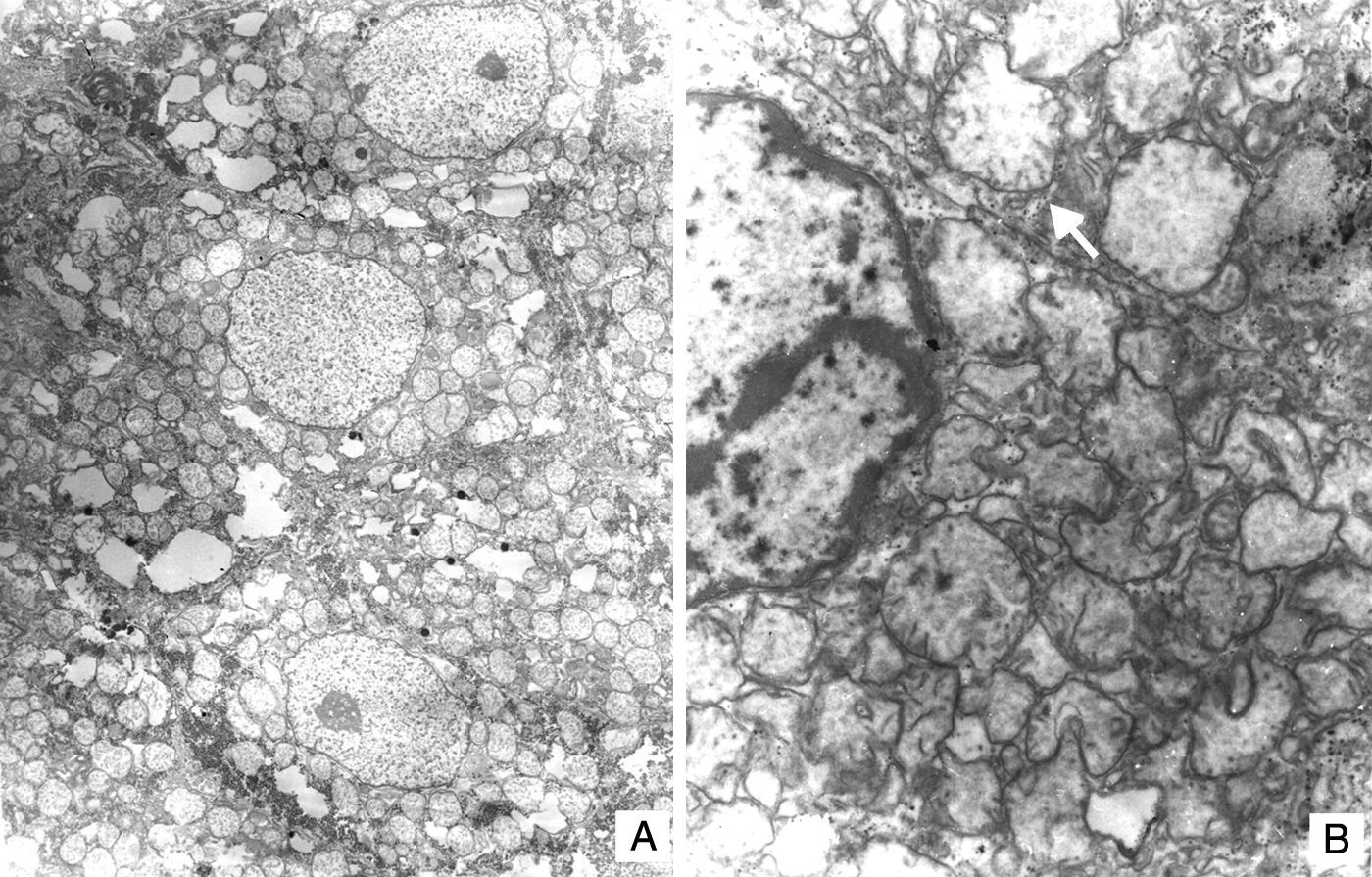
El hígado estaba aumentado de tamaño, con superficie externa micronodular y coloración verde; los cortes seriados mostraron nódulos de regeneración menores de 3mm de diámetro, áreas de necrosis reciente multifocal y dilatación de venas portales intrahepáticas (fig. 3). El estudio histopatológico del hígado mostró una cirrosis micronodular; los nódulos de regeneración estaban rodeados por tabiques delgados de fibrosis y numerosos colangiolos, algunos con tapones de bilis en su luz. Existió necrosis extensa de los hepatocitos, antigua y reciente, de predominio en el centro del lobulillo (centrilobar) asociada con esteatosis macrovesicular y colestasis hepatocelular y canalicular (fig. 4). No se observaron hepatocitos con citoplasmas hipereosinófilos debido al daño intenso y crónico. Los hallazgos fueron concordantes con los descritos por microscopía de luz en el síndrome de depleción del DNA mitocondrial (mtDNA)1. El estudio de inmunohistoquímica con anticuerpos anti-citoqueratina 7 (CK7) reveló la proliferación intensa de canalículos en la periferia de los nódulos de regeneración y la presencia de expresión aberrante de CK7 en el citoplasma de hepatocitos, y realzó la extensa pérdida de hepatocitos (fig. 5). También se observaron depósitos de hierro en hepatocitos y células de Kupffer. El estudio ultraestructural con el microscopio electrónico de transmisión mostró hepatocitos de citoplasma amplio; se observó un aumento en el número de mitocondrias que desplazan el núcleo y los otros organelos. Estas mitocondrias atípicas son de tamaño y forma irregular, con aumento de la matriz de aspecto granular y disminución y marginación a la periferia de las crestas (fig. 6). En otras áreas se observaron depósitos de bilis y cuerpos parecidos a mielina que sugieren el diagnóstico de una enfermedad mitocondrial. Algunos autores consideran estos hallazgos ultraestructurales característicos del síndrome de depleción de mtDNA. Los pacientes con la mutación MPV17 localizada en 2p24.1 presentan cuadros clínicos y hallazgos histopatológicos similares a esta paciente1–4. Sin embargo, sería necesario realizar estudios de biología molecular para demostrar esta mutación.

. B. Hepatocitos con vacuolación del citoplasma y colestasis. Nótese la proliferación de colangiolos en la periferia del nódulo de regeneración (HE 100X).")
 y colangiolos resaltados por expresión citoplasmática de CK7 (IHQ).")
.")
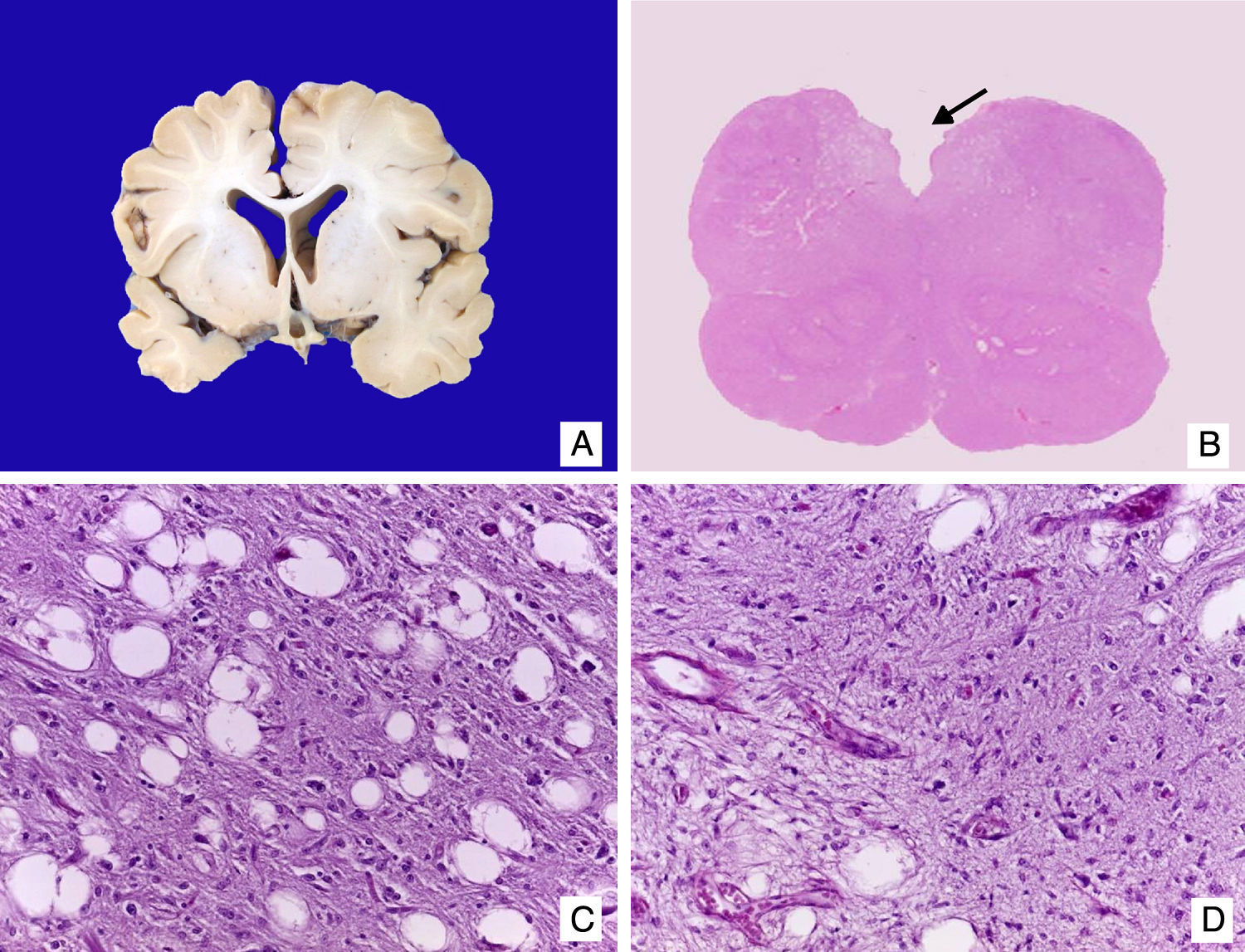
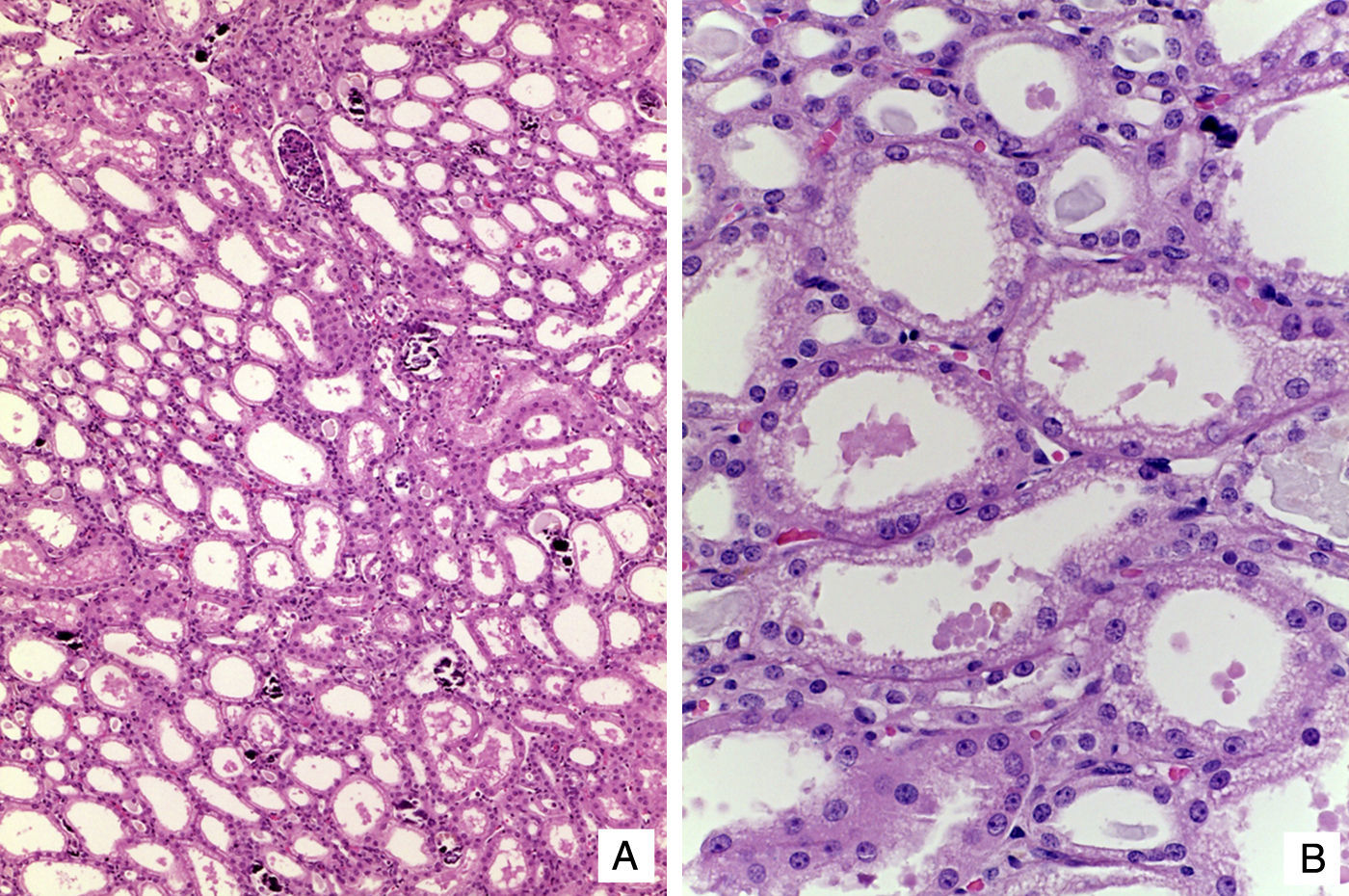
El cerebro mostró datos de atrofia con disminución del peso (750g vs. 810g), surcos profundos y circunvoluciones estrechas, dilatación ex vacuo de los ventrículos laterales y disminución del volumen de la substancia blanca. No se observaron alteraciones en núcleos grises de la base ni cerebelo. El estudio microscópico demostró presencia de astrocitos de Alzheimer tipo 2 en la corteza, que se correlacionan con los datos de insuficiencia hepática en la paciente. Como hallazgo adicional, en el bulbo se observaron áreas de encefalomalacia con proliferación de vasos y vacuolación de la mielina (fig. 7). Los riñones mostraron congestión medular; como hallazgo independiente se observó doble sistema colector en el riñón derecho. Los cortes histológicos mostraron patrón lobular del parénquima, extensa nefrocalcinosis y glomérulos normales. El hallazgo más importante fue la dilatación de los túbulos proximales. Las células epiteliales mostraron en citoplasma grandes vacuolados con borde ciliar conservado. Los cambios ultraestructurales observados fueron similares a los encontrados en las mitocondrias de los hepatocitos que son responsables de la tubulopatía proximal5 (fig. 8). El músculo esquelético fue morfológicamente normal.
. C. Vacuolación del neuropilo y pérdida de mielina. D. Proliferación vascular.")
. B. Túbulos proximales con vacuolación del citoplasma y dilatación de la luz (HE 400X).")
La forma más frecuente del síndrome de depleción del mtDNA es la hepatocerebral, que se presenta en el periodo neonatal, es progresiva y los niños fallecen antes del primer año de edad por insuficiencia hepática (tabla 1). Este síndrome se considera como una alteración en el mantenimiento del mtDNA debido a un defecto en la comunicación entre el DNA nuclear y el mtDNA2.
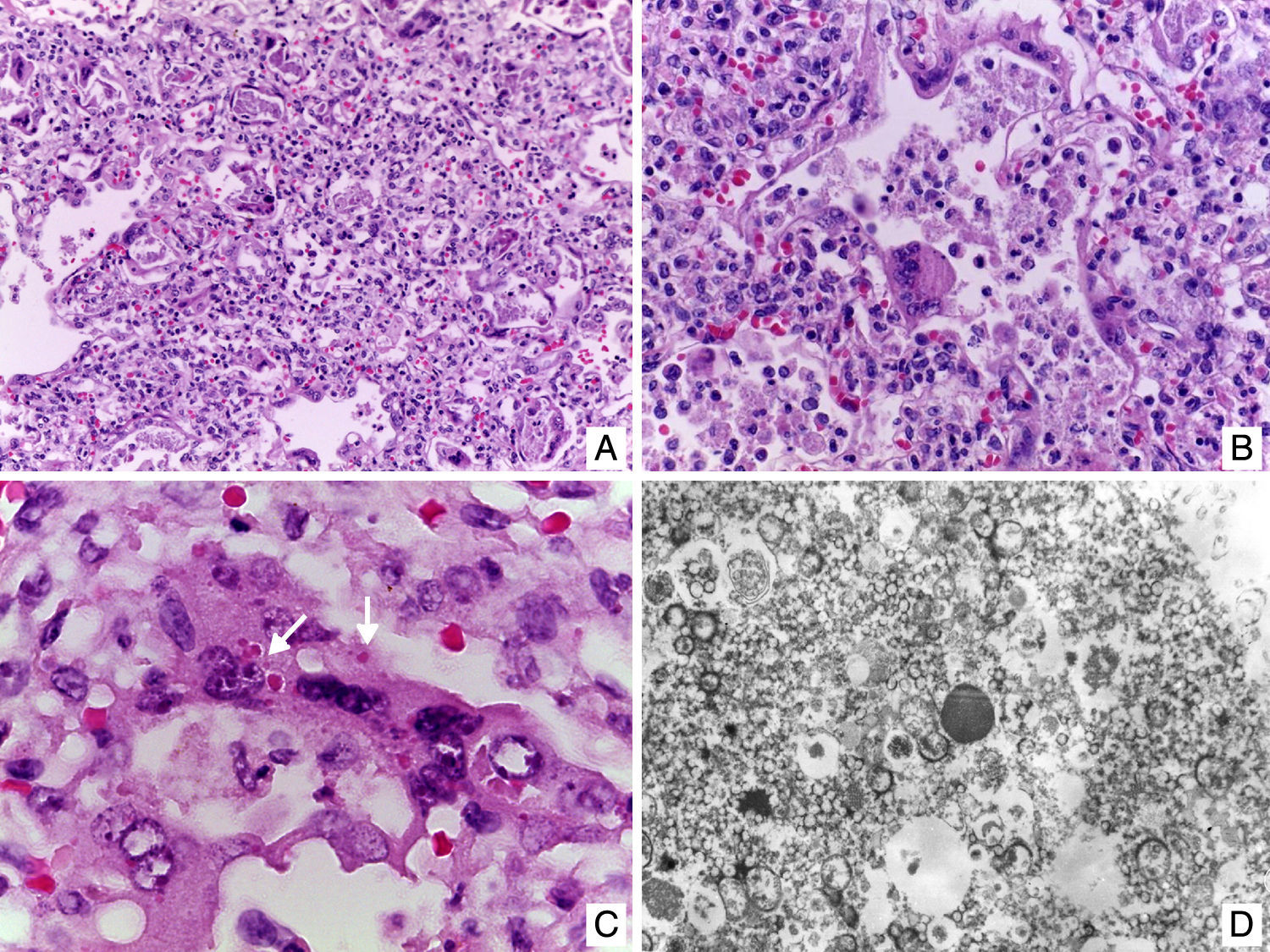
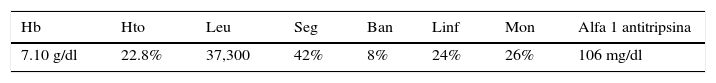
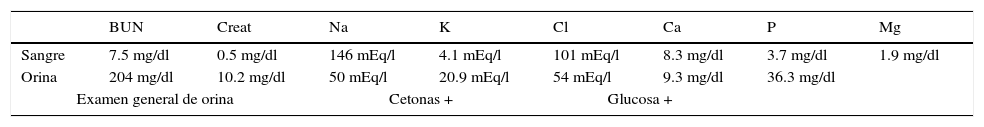
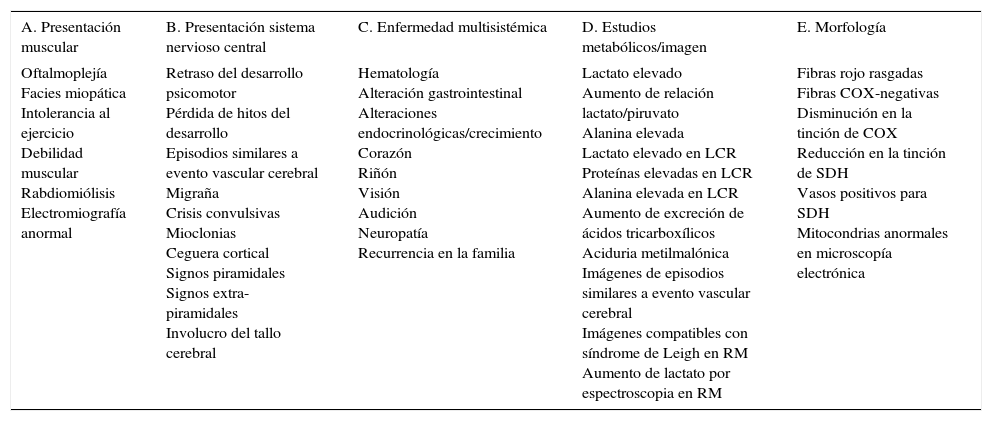
Entre otros hallazgos, se encontró una neumonía viral con daño alveolar difuso con numerosas células gigantes multinucleadas, algunas con inclusiones intracitoplásmicas eosinófilas rodeadas por un halo claro; por microscopía electrónica, las inclusiones se observaron como estructuras electrodensas filamentosas concordantes con virus sincicial respiratorio (fig. 9). El miocardio mostró cardiomiocitos con cambios por miocitólisis colicuativa e hipertrofia, además de datos morfológicos de choque multiorgánico.
. D. Las inclusiones por microscopía electrónica están formadas por estructuras filamentosas concordantes con virus sincicial respiratorio.")
A. Pulmón con datos de neumonía viral. B. Los alveolos se encuentran revestidos por células gigantes multinucleadas y en los tabiques alveolares se observa infiltrado linfocitario. C. Célula gigante multinucleada con inclusiones eosinófilas en el citoplasma (flechas). D. Las inclusiones por microscopía electrónica están formadas por estructuras filamentosas concordantes con virus sincicial respiratorio.
Síndrome de depleción del DNA mitocondrial tipo hepatocerebral con tubulopatía proximal.
4.2Alteraciones concomitantes- •
Hepatomegalia (800g vs. 276g).
- •
Ascitis (950ml).
- •
Cirrosis hepática micronodular.
- •
Colestasis hepatocelular, canalicular y ductal.
- •
Esteatosis hepática macro y microvesicular y hemosiderosis leve.
- •
Alteraciones ultraestructurales mitocondriales en hepatocitos.
- •
Atrofia cerebral (750g vs. 810g).
- •
Encefalomalacia bulbar moderada.
- •
Nefromegalia bilateral.
- •
Tubulopatía proximal con vacuolación del epitelio y alteraciones ultraestructurales mitocondriales.
- •
Nefrocalcinosis extensa.
- •
Neumonía viral de células gigantes por virus sincicial respiratorio por ultraestructura.
- •
Daño alveolar agudo difuso.
- •
Enfermedad vascular pulmonar grado B.
- •
Cardiomegalia (50g vs. 41g).
- •
Miocitólisis colicuativa en miocardio.
- •
Lobulación pulmonar anormal
- •
Doble sistema colector derecho
Esta enfermedad pudo haberse diagnosticado en vida con una biopsia hepática, ya que los hallazgos histopatológicos y ultraestructurales fueron muy sugestivos.
5Comentarios finales5.1Departamento de GenéticaEl genoma humano se puede dividir en dos grandes grupos: el genoma nuclear y el genoma mitocondrial. El genoma nuclear presenta aproximadamente 26 mil genes, mientras que el mitocondrial tiene 37 genes. Trece de los genes mitocondriales codifican para proteínas que componen la cadena respiratoria, el sitio donde se produce ATP a través de la fosforilación oxidativa1. Entre el genoma nuclear y mitocondrial hay una comunicación y una interacción importante. En el DNA nuclear se encuentran genes que codifican para proteínas responsables del mantenimiento, replicación y transcripción del mtDNA, por lo que la adecuada función de ambos genomas es necesaria para el mantenimiento del mtDNA, y por lo tanto para la adecuada producción de ATP6. Las enfermedades mitocondriales representan un grupo de trastornos multisistémicos, que frecuentemente incluyen entre sus manifestaciones la disfunción del sistema nervioso central y periférico, causados por diversas mutaciones localizadas tanto en el genoma mitocondrial como en el nuclear. La prevalencia de estas patologías es de 1 en 5,000 individuos7. Las manifestaciones clínicas de este grupo de patologías pueden darse en cualquier órgano o tejido, a cualquier edad y con cualquier mecanismo de herencia (tabla 4). Debido a la complejidad de estas enfermedades, se han desarrollado criterios diagnósticos o escalas de probabilidad diagnóstica con el objetivo de establecer grados de certeza o posibilidad de la existencia de una disfunción mitocondrial, como la escala de Wolf modificada (tabla 5)8.
Escala de Wolf modificada
| A. Presentación muscular | B. Presentación sistema nervioso central | C. Enfermedad multisistémica | D. Estudios metabólicos/imagen | E. Morfología |
|---|---|---|---|---|
| Oftalmoplejía Facies miopática Intolerancia al ejercicio Debilidad muscular Rabdiomiólisis Electromiografía anormal | Retraso del desarrollo psicomotor Pérdida de hitos del desarrollo Episodios similares a evento vascular cerebral Migraña Crisis convulsivas Mioclonias Ceguera cortical Signos piramidales Signos extra-piramidales Involucro del tallo cerebral | Hematología Alteración gastrointestinal Alteraciones endocrinológicas/crecimiento Corazón Riñón Visión Audición Neuropatía Recurrencia en la familia | Lactato elevado Aumento de relación lactato/piruvato Alanina elevada Lactato elevado en LCR Proteínas elevadas en LCR Alanina elevada en LCR Aumento de excreción de ácidos tricarboxílicos Aciduria metilmalónica Imágenes de episodios similares a evento vascular cerebral Imágenes compatibles con síndrome de Leigh en RM Aumento de lactato por espectroscopia en RM | Fibras rojo rasgadas Fibras COX-negativas Disminución en la tinción de COX Reducción en la tinción de SDH Vasos positivos para SDH Mitocondrias anormales en microscopía electrónica |
LCR: líquido cefalorraquídeo; RM: resonancia magnética; COX: citocromo C oxidasa; SDH: succinato deshidrogenasa.
1 punto = poco probable; 2-4 puntos= posible; 5-7 puntos = probable; 8-12 puntos= definido.
Recientemente se ha aprovechado el avance de técnicas de biología molecular para la detección de mutaciones, como la secuenciación de segunda generación, debido a que mutaciones en mtDNA y nuclear pueden causar signos y síntomas similares9.
El síndrome de depleción de mtDNA es un grupo heterogéneo de alteraciones que se debe a la disminución en el número de copias de mtDNA por mutaciones en genes nucleares que codifican para proteínas encargadas de la replicación y mantenimiento del mtDNA. Tienen un tipo de herencia autosómica recesiva y presentan heterogeneidad genética; es decir, mutaciones en distintos genes pueden causar el síndrome de depleción de mtDNA9,10.
En resumen, ante un neonato o lactante que se presenta con hipoglicemia, hiperlactatemia e hiperbilirrubinemia, se debe considerar la posibilidad de un síndrome de depleción de mtDNA. Con la biopsia hepática se debe considerar la posibilidad de un síndrome de depleción de mtDNA con base en los hallazgos ultraestructurales mitocondriales. Sin embargo, el diagnóstico de certeza se debe realizar por biología molecular.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.