


El síndrome de Silver-Russell presenta restricción del crecimiento intrauterino y posnatal, macrocefalia relativa y asimetría, entre otras características. Es causado por mecanismos genéticos y epigenéticos en el cromosoma 11p15.5 en el 40% de los casos y por disomía uniparental materna del cromosoma 7 en el 10%.
MétodosSe identificaron los pacientes con diagnóstico de síndrome de Silver-Russell del Hospital Infantil de México Federico Gómez atendidos de 1998 a 2012; se reevaluaron 20 pacientes según los criterios clínicos internacionales, y se confirmó el diagnóstico en nueve sujetos.
ResultadosTodos los pacientes presentaron restricción del crecimiento intrauterino y talla baja, ambos criterios diagnósticos mayores. La macrocefalia relativa estuvo presente en el 78% y la asimetría corporal solo en el 33%. Otras características, como la acidosis tubular renal, estuvieron presentes en más del 50%.
ConclusionesEl diagnóstico del síndrome de Silver-Russell es complejo, por lo que contar con criterios clínicos adecuados es fundamental. Dado que la talla baja es la principal solicitud de atención médica en este síndrome, es relevante establecer diagnósticos diferenciales y valorar el crecimiento y desarrollo de todos los pacientes para identificar a aquellos en quienes la talla baja forma parte de una entidad sindrómica y que serían candidatos para realizar estudios moleculares. Este abordaje tendrá implicaciones para su manejo, pronóstico y asesoramiento genético.
Patients with Silver-Russell syndrome suffer from severe intrauterine and postnatal growth retardation, relative macrocephaly and body asymmetry, among other characteristics. It is caused by several genetic and epigenetic mechanisms in 11p15.5 in 40% of the cases and maternal uniparental disomy of chromosome 7 in 10%.
MethodsTwenty patients with a diagnosis of Silver-Russell syndrome who were seen at the HIMFG from 1998 to 2012, were evaluated according to international clinical criteria confirming the diagnosis in nine of the subjects.
ResultsAll patients showed intrauterine and postnatal growth retardation and short stature, both considered as major criteria of Silver-Russell syndrome. Relative macrocephaly was present in 78% of the patients and asymmetry in 33%. Other characteristics such as renal tubular acidosis were present > 50% of the cases.
ConclusionsThe clinical diagnosis of Silver-Russell syndrome is complex. Short stature is the main reason for seeking medical attention and is helpful in the identification of a differential diagnosis. This situation underlines the importance of growth and development evaluation of all patients and particularly in those with short stature to identify those cases that may require molecular studies, with implications in management, prognosis and genetic counseling.
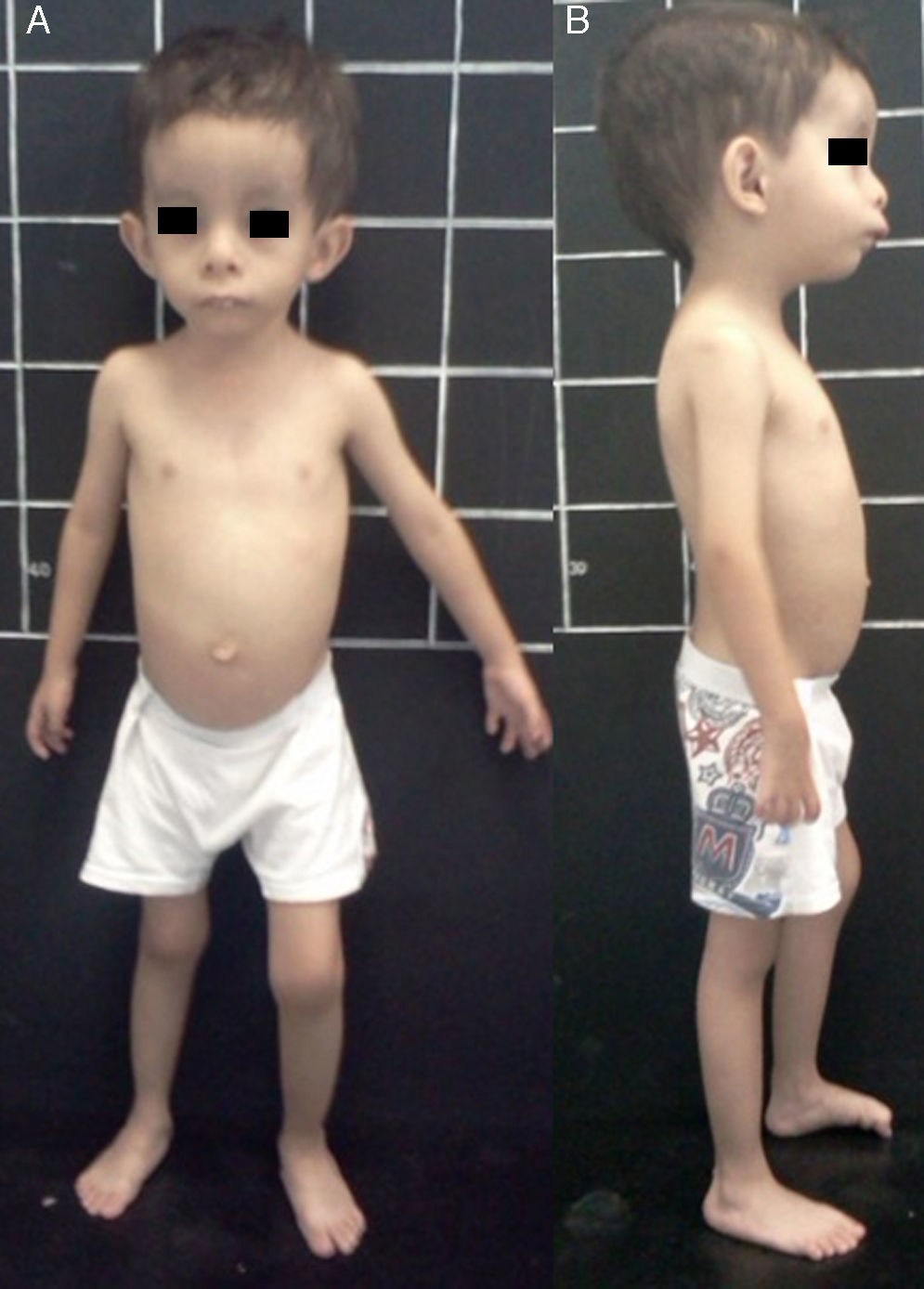
El síndrome de Silver-Russell (SSR) (MIM 180860) se caracteriza por restricción del crecimiento intrauterino (RCIU) y posnatal, macrocefalia relativa, cara triangular, asimetría facial y/o corporal y por alteraciones en la alimentación (fig. 1)1. Fue descrito por primera vez en 1953 por el pediatra estadounidense Henry Silver, quien reportó dos casos de niños con peso bajo al nacer, restricción del crecimiento posnatal y asimetría corporal2. Sin conocimiento previo de ello, en 1954 el inglés Alexander Russell describió 5 pacientes con características similares3.
 Vista de frente. B) Vista de perfil. Nótese asimetría hemicorporal, macrocefalia relativa, facies triangular, frontal prominente y retromicrognatia.")
La incidencia de SSR es de 1:3,000 a 1:10,000 recién nacidos vivos y ocurre en todos los grupos étnicos sin predilección de sexo4. Además de las características clínicas previamente mencionadas, los pacientes con SSR presentan hipotonía e hipotrofia muscular, manchas café con leche, cierre tardío de fontanelas, frontal prominente, comisuras labiales descendentes, micro/retrognatia, alteraciones en el espacio interdental, pabellones auriculares dismórficos y de implantación baja, criptorquidia, hipospadias, camptodactilia, contracturas articulares, clinodactilia del quinto dedo, sindactilia entre segundo y tercer ortejo, sudoración excesiva, hipoglucemia, voz aguda, retraso en el desarrollo psicomotor y enfermedad por reflujo gastroesofágico1,4,5 (fig. 1). En el SSR se observa expresividad variable con un espectro clínico que puede ir desde el cuadro clínico clásico hasta manifestaciones mínimas que, incluso, pueden pasar desapercibidas. Lo anterior, aunado a la dificultad para definir las características clínicas principales en esta entidad, hacen que su diagnóstico clínico sea difícil, y por ende se considere que está subdiagnosticado5.
Si bien la mayoría de los casos de SSR tienen una presentación esporádica, la participación de factores genéticos en su etiología ha sido demostrada. Existen reportes de presentaciones familiares sugiriendo diferentes patrones de herencia: autosómico dominante o recesivo6 (y de manera anecdótica, ligado al X [MIM 312780]). También se han identificado casos familiares asociados con diversas aberraciones cromosómicas, entre ellas rearreglos numéricos y estructurales que implican de forma recurrente los cromosomas 7, 11p y 17q7,8.
Inicialmente se propuso como base etiológica del SSR una alteración o estrés intrauterino a las 6-7 semanas de gestación9; otros autores consideraron tanto la falta de respuesta de un órgano blanco a la hormona de crecimiento como una alteración estructural en su molécula4,9. Actualmente se considera que el SSR presenta heterogeneidad genética debido a que diversos mecanismos genéticos y epigenéticos están implicados en su etiología, principalmente por alteraciones en la impronta10. En el genoma humano existe un grupo de genes improntados; es decir, genes que presentan una expresión diferencial de sus alelos dependiendo del progenitor del cual se han heredado. Los genes improntados se encuentran involucrados en diversos aspectos del crecimiento y comportamiento, por lo que varios síndromes asociados con alteraciones en la impronta se caracterizan clínicamente por este tipo de alteraciones11. Con relación al SSR, los mecanismos que pueden afectar la impronta son diversos y están asociados con alteraciones que modifican los patrones de metilación en la región 11p15.5, en el 40-50% de los casos, y a disomía uniparental (DUP) materna del cromosoma 7 —DUP(7)mat—, en el 10%1,12.
La impronta genómica es un proceso epigenético mediante el cual en las líneas germinales materna y paterna se confieren marcas particulares a grupos de genes localizados en diferentes regiones cromosómicas específicas que se heredarán en los gametos. Las marcas epigenéticas incluyen diferencias en la metilación de dinucleótidos CpG localizados en secuencias reguladoras de los genes o regiones cromosómicas improntadas, llamadas centros de regulación de la impronta (CRI) y modificaciones en las histonas que cambian la estructura de la cromatina. Estas modificaciones son estables durante la división mitótica y mantenidas durante el desarrollo y crecimiento del individuo. Las diferencias epigenéticas entre las copias materna o paterna de los alelos improntados permiten la expresión génica de un solo alelo, mientras que reprimen la expresión del otro, por lo que las alteraciones en el establecimiento o mantenimiento de la impronta pueden llevar a la sobreexpresión o falta de los productos de los genes implicados13.
El término DUP se refiere a la presencia de ambos cromosomas homólogos heredados de un mismo progenitor. La DUP puede presentar dos variantes: isodisomía, cuando solo uno de los cromosomas homólogos se encuentra por duplicado, y heterodisomía, cuando se heredan ambos cromosomas homólogos de un solo progenitor. La DUP puede ser ocasionada por diversos mecanismos, entre ellos el rescate monosómico, ya sea por fertilización de un gameto nulisómico o por un error en la disyunción mitótica; eventos de recombinación mitótica; el rescate trisómico, o la complementación de gametos. La DUP de cromosomas o regiones con genes improntados modifica la dosis génica funcional, ocasionando alteraciones en el crecimiento y desarrollo14.
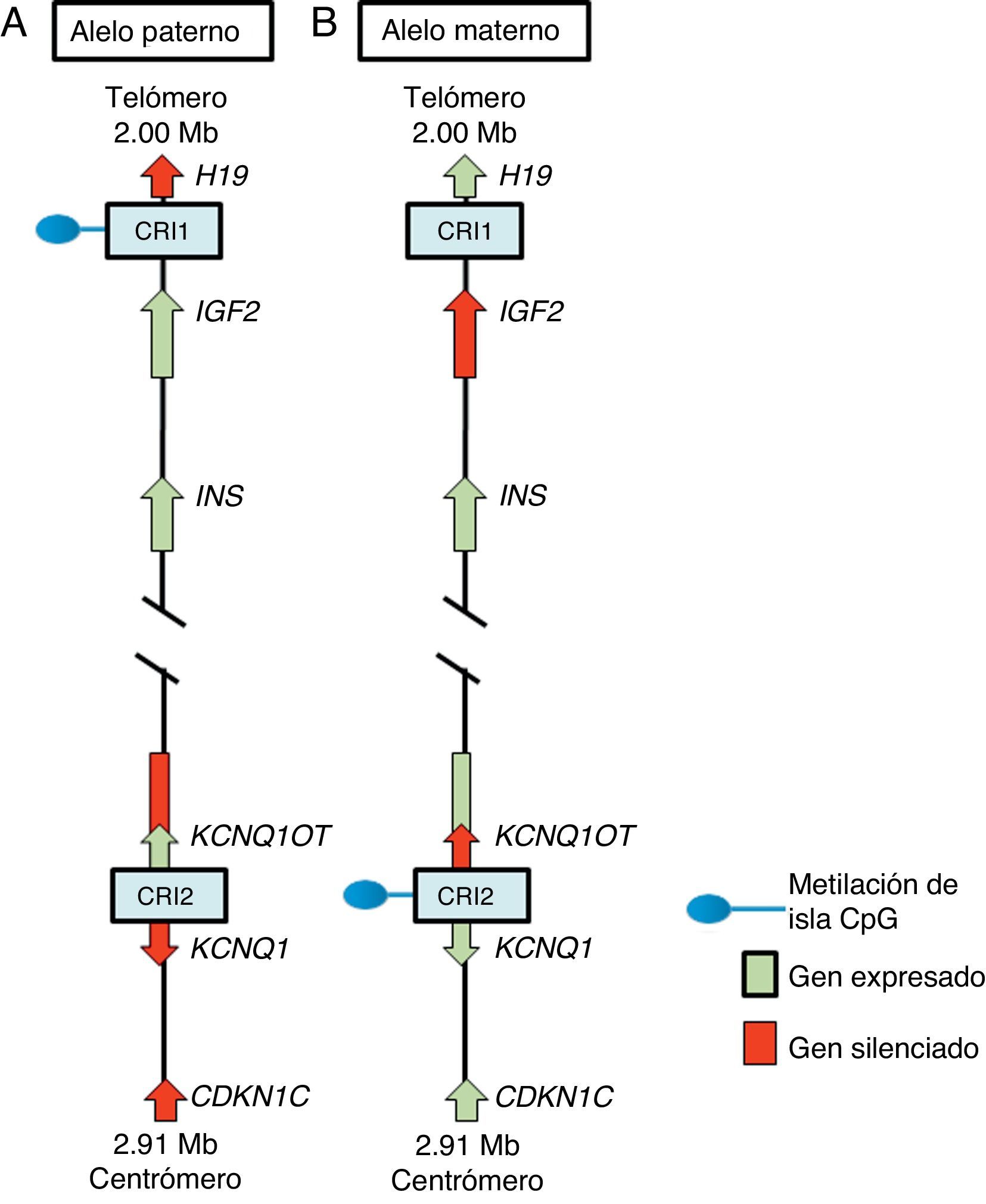
En la región 11p15.5 se encuentra una agrupación de genes improntados relevante para el SSR12 (fig. 2), los cuales desempeñan un papel importante en el control del crecimiento fetal15. Esta región cromosómica se organiza en dos diferentes dominios o regiones improntados, cada uno bajo el control de su propio CRI que actúa en cis, es decir, que regula la expresión de los genes adyacentes. A los CRI de la región 11p15.5 se les conoce como CRI1 o telomérico y CRI2 o centromérico y tienen mecanismos reguladores diferentes; CRI1 funciona como un aislante, mientras que CRI2 es el promotor de un macro ncRNA16. Estos CRI tienen patrones de metilación opuestos; CRI1 se encuentra metilado en los gametos masculinos, mientras que CRI2 es metilado en la línea germinal materna1 (fig. 2). El CRI1 controla la expresión monoalélica de dos genes ampliamente estudiados: IGF2, con expresión del alelo paterno y que codifica para el factor de crecimiento similar a la insulina 2, y el gen H19, con expresión del alelo materno y que codifica para un long intergenic non coding RNA (lincRNA)17. Los genes H19 e IGF2 se expresan ampliamente durante el desarrollo embrionario, y disminuyen posnatalmente en casi todos los tejidos. El IGF2 desempeña un papel principal como promotor del crecimiento placentario y embrionario, mientras que la función exacta de H19 es incierta. La expresión de H19 e IGF2 es regulada por la unión de la proteína CTCF en el CRI1 hipometilado del alelo materno. Las proteínas cohesinas contribuyen a la regulación génica de los sitios de unión de CTCF en CRI1 mediante la formación de bucles de cromatina que permiten la expresión de uno u otro gen16.
 Cromosoma paterno y (B) cromosoma materno mostrando las diferencias en los patrones de metilación de las regiones de control de la impronta (CRI1 y CRI2) y en la expresión de los alelos de los genes H19, IGF2, KCNQ1, KCNQ1OT y CDKN1C.")
Región improntada en el cromosoma 11p15.5. (A) Cromosoma paterno y (B) cromosoma materno mostrando las diferencias en los patrones de metilación de las regiones de control de la impronta (CRI1 y CRI2) y en la expresión de los alelos de los genes H19, IGF2, KCNQ1, KCNQ1OT y CDKN1C.
El CRI2, que incluye a KvDMR1, regula la expresión de los genes KCNQ1, KCNQ1OT1 y CDKN1C. En individuos normales, el CRI2 se encuentra metilado en el alelo materno y resulta en la supresión de KCNQ1OT1 y en la expresión de KCNQ1 y, particularmente, de CDKN1C, que es un regulador negativo de la proliferación y crecimiento celular que participa en el desarrollo fetal humano, ya que codifica para un inhibidor de los complejos ciclina-cinasa18. El RNA KCNQ1OT1 es expresado solo del alelo paterno y resulta en el silenciamiento de todos los genes improntados en el dominio, incluyendo CDKN1C. En el CRI2 hipometilado en el alelo paterno se ha identificado también la unión de CTCF16.
El síndrome de Beckwith-Wiedemann (MIM 130650) fue la primera enfermedad en la que se encontraron alteraciones en la impronta de la región 11p15.5, y cuando se consideran las manifestaciones clínicas, es particularmente interesante que mientras el SSR se caracteriza por RCIU y talla baja, el síndrome de Beckwith-Wiedemann se considera un síndrome de sobrecrecimiento19. En el 40-50% de los casos de SSR se encuentra hipometilación en el CRI1 del cromosoma 11p15.5 paterno, lo que lleva a la sobreexpresión del gen H19 y a la disminución de la expresión del promotor de crecimiento IGF21. En algunos casos el SSR se debe a duplicaciones del alelo materno o a casos de DUP materna segmentaria del cromosoma 11 con aumento de la expresión de los genes KCNQ1 y CDKN1C16,18. El CRI2 se encuentra raramente afectado por epimutaciones en SSR, mientras que la hipometilación del alelo materno de esta región diferencialmente metilada es la principal causa de síndrome de Beckwith-Wiedemann18,19.
Por otra parte, la DUP(7)mat ha sido comprobada en el 10% de los casos con SSR y se ha relacionado con alteraciones en la expresión de genes improntados en el cromosoma 7, por lo que se están investigando posibles regiones y genes candidatos en dicho cromosoma8,14. En particular se han identificado genes improntados en 7p11.2-p13 y 7q31-q34. Cada uno de estos loci contiene, como mínimo, un gen candidato que participa en la regulación del crecimiento; entre ellos se encuentran el gen GRB10, el cual es expresado del alelo materno y codifica para la proteína 10 de unión a receptor de factor de crecimiento, y el gen PEG1/MEST, el cual se expresa en el alelo paterno y codifica para el transcrito expresado en el mesodermo10. El fenotipo de los pacientes con SSR presenta diferencias dependiendo de su etiología; así, los pacientes con hipometilación del CRI1 de 11p15.5 presentan el fenotipo «clásico» de SSR, además de una mayor incidencia de asimetría corporal en comparación con los pacientes cuya etiología es debida a DUP(7)mat7,20.
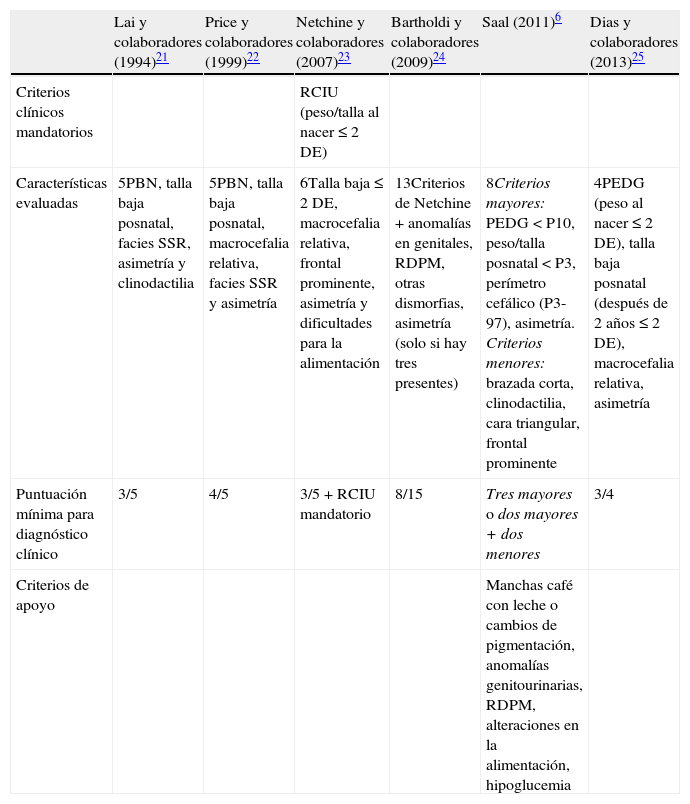
Debido a que las bases etiológicas del SSR son complejas, el diagnóstico molecular es complicado, y en la mitad de los casos, no concluyente. Por ello, es necesario basarse en un diagnóstico clínico de certeza; sin embargo, este continúa siendo un reto por la variabilidad en la expresión fenotípica. A la fecha se han propuesto varios sistemas de evaluación basados en puntaje para delinearlo (tabla 1)6,21–25 sin que se cuente con un sistema de puntuación o criterios diagnósticos aceptados universalmente26,27. Aunado a lo anterior, las características faciales tienden a atenuarse en la infancia tardía y aún más en la edad adulta1,25; por ello el SSR es muchas veces considerado como un diagnóstico presuntivo.
Criterios diagnósticos propuestos para SSR
| Lai y colaboradores (1994)21 | Price y colaboradores (1999)22 | Netchine y colaboradores (2007)23 | Bartholdi y colaboradores (2009)24 | Saal (2011)6 | Dias y colaboradores (2013)25 | |
| Criterios clínicos mandatorios | RCIU (peso/talla al nacer≤ 2DE) | |||||
| Características evaluadas | 5PBN, talla baja posnatal, facies SSR, asimetría y clinodactilia | 5PBN, talla baja posnatal, macrocefalia relativa, facies SSR y asimetría | 6Talla baja≤ 2DE, macrocefalia relativa, frontal prominente, asimetría y dificultades para la alimentación | 13Criterios de Netchine+anomalías en genitales, RDPM, otras dismorfias, asimetría (solo si hay tres presentes) | 8Criterios mayores: PEDG<P10, peso/talla posnatal<P3, perímetro cefálico (P3-97), asimetría. Criterios menores: brazada corta, clinodactilia, cara triangular, frontal prominente | 4PEDG (peso al nacer≤ 2DE), talla baja posnatal (después de 2 años≤ 2DE), macrocefalia relativa, asimetría |
| Puntuación mínima para diagnóstico clínico | 3/5 | 4/5 | 3/5+RCIU mandatorio | 8/15 | Tres mayores o dos mayores+dos menores | 3/4 |
| Criterios de apoyo | Manchas café con leche o cambios de pigmentación, anomalías genitourinarias, RDPM, alteraciones en la alimentación, hipoglucemia |
DE: desviación estándar; PBN: peso bajo al nacer; PEDG: pequeño para la edad gestacional; RCIU: restricción del crecimiento intrauterino; RDPM: retraso en el desarrollo psicomotor. SSR: síndrome de Silver-Russell.
El diagnóstico diferencial de SSR incluye cualquier entidad que involucre RCIU y talla baja, y deben descartarse síndromes secundarios a aberraciones cromosómicas y alteraciones en la reparación del DNA, así como los síndromes 3-M (MIM 273750), Dubowitz (MIM 223370), IMAGe (MIM 614732) y el síndrome por exposición del feto al alcohol9. Todos estos aspectos, a su vez, complican el poder otorgar un asesoramiento genético de certeza.
En este trabajo se describen las características clínicas de una serie de pacientes con diagnóstico clínico de SSR atendidos en el Departamento de Genética del Hospital Infantil de México Federico Gómez de 1998 a 2012. El presente trabajo es parte del protocolo de investigación «Implicaciones del patrón de metilación en la región 11p15.5 como mecanismo etiológico de la hemihiperplasia aislada» (HIM/2012/007). Como ha sido descrito previamente por Moreno y colaboradores28, en este protocolo se incluyeron como controles para estudio molecular de la región 11p15.5 pacientes con diagnóstico clínico de SSR; por ello se realizó la revisión del archivo clínico, la reevaluación clínica de los pacientes identificados y, finalmente, se llevó a cabo el análisis estadístico de las características presentes en los pacientes con criterios diagnósticos compatibles con SSR. Se hizo un análisis comparativo de los pacientes utilizando los diferentes criterios diagnósticos propuestos para esta entidad (tabla 1). El estudio molecular de estos casos será reportado en el contexto del protocolo realizado.
2MétodosEl universo de estudio incluyó a pacientes registrados con diagnóstico clínico de SSR en el Departamento de Bioestadística y Archivo Clínico de nuestra Institución de 1998 a 2012. Se realizó un estudio descriptivo y retrospectivo. Se identificaron 20 pacientes quienes fueron reevaluados clínicamente de acuerdo con los criterios propuestos por Saal6 para confirmar o descartar el diagnóstico clínico de SSR. Los criterios de Saal6 se basan en la evaluación de la presencia de ocho características clínicas, divididas en criterios mayores y menores. Ante la presencia de tres características de criterio mayor o dos de criterio mayor y dos de criterio menor, se considera el diagnóstico de SSR. Además, la escala analiza cinco características más para apoyar el diagnóstico (tabla 1). Los pacientes que no cumplieron con dichos criterios fueron excluidos de este análisis.
3ResultadosDel total de 20 pacientes identificados por registro con diagnóstico SSR, solo nueve cumplieron con los criterios clínicos propuestos por Saal6 para SSR. De los 11 pacientes excluidos, tres fueron diagnosticados como talla baja familiar, tres como talla baja secundaria a acidosis tubular renal (ATR) y uno como síndrome de Dubowitz; los cuatro pacientes restantes continúan en estudio.
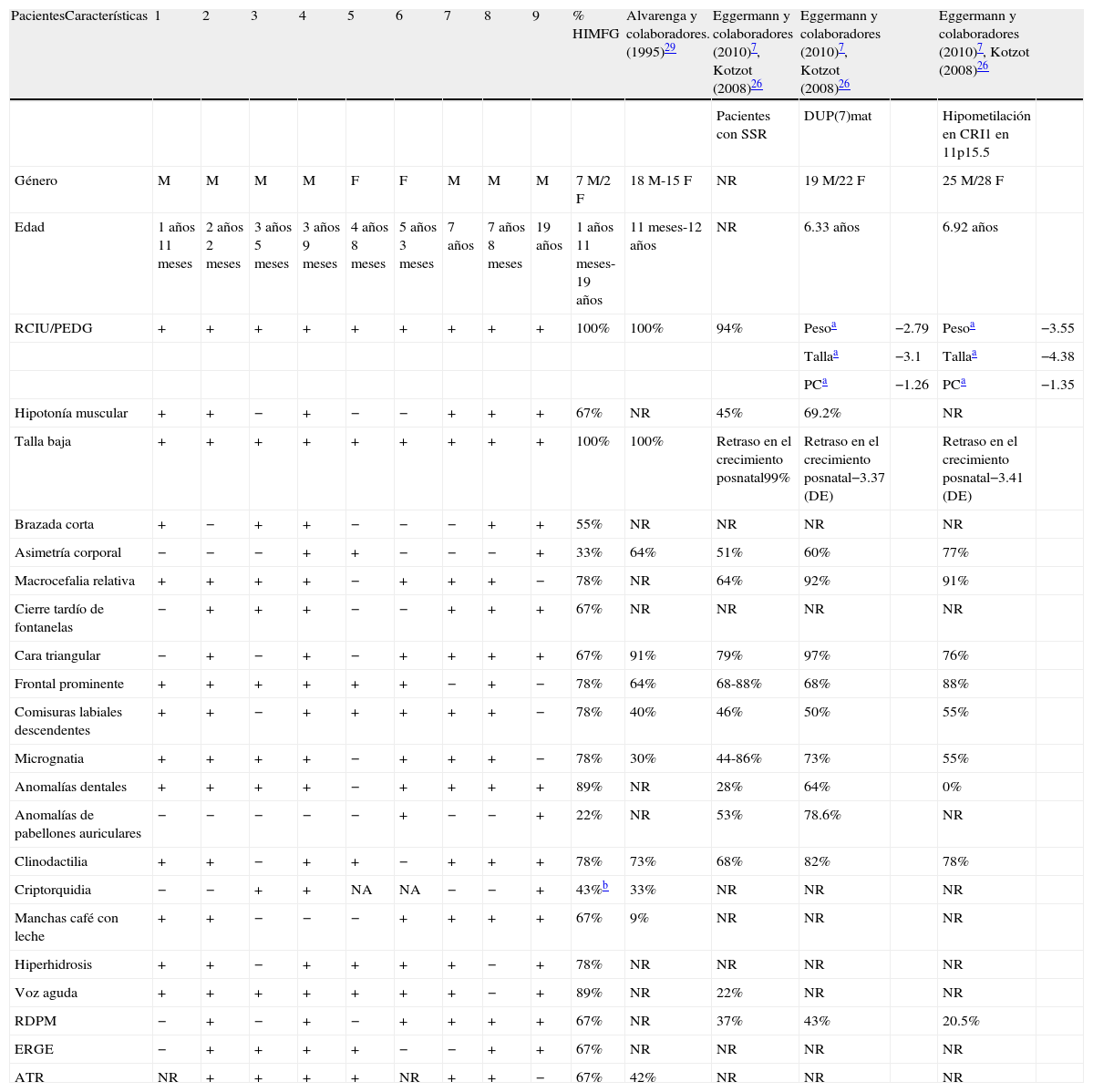
Con relación al análisis de las características clínicas de los nueve pacientes con diagnóstico clínico de SSR, siete fueron masculinos y dos femeninos, con un rango de edad entre un año 11 meses y 19 años. El resultado de cariotipo fue normal en los cinco pacientes (pacientes 2, 4, 6, 7 y 9) estudiados citogenéticamente. Las características clínicas de los pacientes y los porcentajes en que se encontraron en esta serie se muestran en la tabla 226,27,29.
Comparación clínica de los pacientes con diagnóstico clínico de SSR y algunas series reportadas en la literatura
| PacientesCaracterísticas | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | % HIMFG | Alvarenga y colaboradores. (1995)29 | Eggermann y colaboradores (2010)7, Kotzot (2008)26 | Eggermann y colaboradores (2010)7, Kotzot (2008)26 | Eggermann y colaboradores (2010)7, Kotzot (2008)26 | ||
| Pacientes con SSR | DUP(7)mat | Hipometilación en CRI1 en 11p15.5 | ||||||||||||||
| Género | M | M | M | M | F | F | M | M | M | 7M/2F | 18M-15F | NR | 19M/22F | 25M/28F | ||
| Edad | 1años 11meses | 2años 2meses | 3años 5meses | 3años 9meses | 4años 8meses | 5años 3meses | 7años | 7años 8meses | 19 años | 1años 11meses-19años | 11meses-12años | NR | 6.33 años | 6.92 años | ||
| RCIU/PEDG | + | + | + | + | + | + | + | + | + | 100% | 100% | 94% | Pesoa | −2.79 | Pesoa | −3.55 |
| Tallaa | −3.1 | Tallaa | −4.38 | |||||||||||||
| PCa | −1.26 | PCa | −1.35 | |||||||||||||
| Hipotonía muscular | + | + | − | + | − | − | + | + | + | 67% | NR | 45% | 69.2% | NR | ||
| Talla baja | + | + | + | + | + | + | + | + | + | 100% | 100% | Retraso en el crecimiento posnatal99% | Retraso en el crecimiento posnatal−3.37 (DE) | Retraso en el crecimiento posnatal−3.41 (DE) | ||
| Brazada corta | + | − | + | + | − | − | − | + | + | 55% | NR | NR | NR | NR | ||
| Asimetría corporal | − | − | − | + | + | − | − | − | + | 33% | 64% | 51% | 60% | 77% | ||
| Macrocefalia relativa | + | + | + | + | − | + | + | + | − | 78% | NR | 64% | 92% | 91% | ||
| Cierre tardío de fontanelas | − | + | + | + | − | − | + | + | + | 67% | NR | NR | NR | NR | ||
| Cara triangular | − | + | − | + | − | + | + | + | + | 67% | 91% | 79% | 97% | 76% | ||
| Frontal prominente | + | + | + | + | + | + | − | + | − | 78% | 64% | 68-88% | 68% | 88% | ||
| Comisuras labiales descendentes | + | + | − | + | + | + | + | + | − | 78% | 40% | 46% | 50% | 55% | ||
| Micrognatia | + | + | + | + | − | + | + | + | − | 78% | 30% | 44-86% | 73% | 55% | ||
| Anomalías dentales | + | + | + | + | − | + | + | + | + | 89% | NR | 28% | 64% | 0% | ||
| Anomalías de pabellones auriculares | − | − | − | − | − | + | − | − | + | 22% | NR | 53% | 78.6% | NR | ||
| Clinodactilia | + | + | − | + | + | − | + | + | + | 78% | 73% | 68% | 82% | 78% | ||
| Criptorquidia | − | − | + | + | NA | NA | − | − | + | 43%b | 33% | NR | NR | NR | ||
| Manchas café con leche | + | + | − | − | − | + | + | + | + | 67% | 9% | NR | NR | NR | ||
| Hiperhidrosis | + | + | − | + | + | + | + | − | + | 78% | NR | NR | NR | NR | ||
| Voz aguda | + | + | + | + | + | + | + | − | + | 89% | NR | 22% | NR | NR | ||
| RDPM | − | + | − | + | − | + | + | + | + | 67% | NR | 37% | 43% | 20.5% | ||
| ERGE | − | + | + | + | + | − | − | + | + | 67% | NR | NR | NR | NR | ||
| ATR | NR | + | + | + | + | NR | + | + | − | 67% | 42% | NR | NR | NR |
ATR: acidosis tubular renal; CRI1: centro de regulación de la impronta de la región 11p15.5 o telomérico; DE: desviaciones estándar; DUP(7)mat: disomía uniparental materna del cromosoma 7; ERGE: enfermedad por reflujo gastroesofágico; F: femenino; HIMFG: Hospital Infantil de México Federico Gómez; M: masculino; NA: no aplica; NR: no reportado; PC: perímetro cefálico; PEDG: pequeño para la edad gestacional; RDPM: retraso en el desarrollo psicomotor; SSR: síndrome de Silver-Russell; RCIU: retraso en el crecimiento intrauterino.
La evaluación de los 20 pacientes registrados con diagnóstico de SSR utilizando los criterios de Saal6 permitió descartar dicho diagnóstico en 11 de ellos; en 7/11 (64%) se integró un diagnóstico diferente. De los nueve pacientes (el 45% del total registrado) en quienes se confirmó el diagnóstico de SSR, hubo un mayor porcentaje de pacientes de sexo masculino (77.77%); este dato llama la atención debido a que se considera que el SSR no presenta predilección por ningún sexo4. En esta serie, el 43% de los pacientes masculinos presentó criptorquidia uni o bilateral (tabla 2). En la serie de Alvarenga y colaboradores29 la criptorquidia estuvo presente en el 33% de sus pacientes; la frecuencia elevada de esta característica podría influir en la atención temprana de un mayor número de pacientes masculinos con SSR, debido a que es un dato de alarma para recibir atención quirúrgica, independientemente de que otros datos del SSR pudiesen estar presentes.
El SSR es una entidad de difícil diagnóstico debido a la gran variabilidad en su expresión y a la poca especificidad de sus manifestaciones principales. La talla baja es el signo cardinal por el que la mayoría de los pacientes con SSR solicitan atención médica; en esta serie todos los pacientes en quienes se confirmó el diagnóstico la presentaron. Este dato resulta igualmente importante para considerar los diagnósticos diferenciales con similitud fenotípica. La talla baja se define como aquella ≤2 desviaciones estándar de la media para la edad27,30; sus causas más comunes son la talla baja familiar y el retraso en el crecimiento constitucional, que en conjunto suman hasta el 75%. Otras etiologías son enfermedades crónicas (10%), entidades sindrómicas (6%), cromosomopatías (5%), displasias óseas (1%) y deficiencia de hormona de crecimiento o insensibilidad a su receptor (1-2%), así como un aporte inadecuado de nutrientes y problemas psicológicos31; por ello es importante descartar las causas mencionadas, que son mucho más frecuentes, antes de considerar el diagnóstico de SSR.
Ante un paciente con talla baja de causa desconocida, debe realizarse una somatometría estricta de los segmentos corporales, valorar la velocidad de crecimiento y comparar la talla del paciente con la talla blanco familiar30. Además, deben analizarse los niveles de electrólitos, concentración de urea, función tiroidea, biometría hemática completa, análisis urinario, anticuerpos antigliandina, edad ósea y las pruebas endocrinológicas adicionales que se requieran según el caso. Debe considerarse también el análisis citogenético, por ejemplo para descartar síndrome de Turner o en pacientes con retraso mental con o sin malformaciones o dismorfias. En los casos de talla baja familiar, la maduración ósea es consistente con la edad cronológica, y la talla final debe estimarse con la talla blanco familiar; en aquellos casos con retraso constitucional del crecimiento y desarrollo, la maduración ósea es consistente con la edad para la estatura, y el pronóstico para una talla adulta normal es bueno30–32. En el SSR la talla baja es proporcionada, si bien puede existir asimetría corporal; de hecho se presenta RCIU, lo cual se manifiesta al nacimiento como una talla menor a la esperada, y este patrón se mantiene durante el desarrollo subsecuente, por debajo de 2 desviaciones estándar.
Dentro de los principales síndromes asociados con talla baja e historia de peso bajo al nacer se encuentra el síndrome de Dubowitz, entidad autosómica recesiva de la que han sido descritos aproximadamente 200 casos y que no tiene predilección étnica o de sexo33. Esta patología es el principal diagnóstico diferencial del SSR ya que comparten varias características clínicas: RCIU, retraso en el crecimiento posnatal, micrognatia, apiñamiento dental, anomalías de pabellones auriculares, alteraciones genitourinarias y voz aguda. Adicionalmente presenta cara triangular, asimetría facial, ptosis (65%) y alteraciones genitourinarias33,34. En este estudio, en uno de los pacientes en quien se descartó SSR, se integró el diagnóstico de síndrome de Dubowitz, ya que el paciente solo contaba con dos criterios mayores para SSR (peso bajo para la edad gestacional, peso y talla bajos posnatales<P3) y presentó además datos no consistentes con SSR y sí asociados con el síndrome de Dubowitz, como microcefalia, bicitopenia, infecciones en vías urinarias recurrentes y alergias alimentarias, además de un fenotipo facial compatible.
En tres de los pacientes excluidos en esta serie se diagnosticó talla baja no menor a la P3 y secundaria a ATR. La ATR por sí sola puede ser una causa de talla baja no sindrómica; en estos pacientes no se encontraron más datos asociados con SSR. Cabe señalar que la ATR estuvo presente en el 67% de los pacientes de esta serie y en el 42% de los pacientes descritos por Alvarenga y colaboradores29; esto lleva a considerar si la ATR es una característica del SSR, ya que hasta ahora no se incluye como uno de los criterios diagnósticos.
Los porcentajes de las características clínicas identificadas en el grupo de pacientes con SSR fueron consistentes con los datos publicados5,7,29 (tabla 2). Como ya se mencionó, todos los pacientes presentaron RCIU y talla baja, datos clínicos cardinales en todos los criterios diagnósticos propuestos (tabla 1). Utilizando la propuesta de Saal6, todos los pacientes tuvieron al menos tres criterios mayores y solo el paciente 4 presentó los cuatro criterios mayores. De los otros criterios mayores, el 78% de nuestros pacientes presentó macrocefalia relativa y solo el 33% asimetría corporal, en contraste con lo reportado en otras series del 64% y el 51%, respectivamente5,7,26. Llamó la atención que la brazada corta se encontró en más de la mitad de estos pacientes, ya que, si bien este es un criterio menor, no está reportado en porcentajes en otras poblaciones; sin embargo, su alta frecuencia en esta población apoyaría su uso como un criterio mayor para el SSR. La alta frecuencia de las características señaladas como criterios menores, tanto en nuestra población como en las reportadas en la literatura, apoyaría el uso de estas características para el diagnóstico del SSR5,7,26. Entre los criterios diagnósticos de apoyo que tuvieron una frecuencia elevada de presentación en los pacientes de esta serie se encuentran las manchas café con leche, la enfermedad por reflujo gastroesofágico, el retraso en el desarrollo psicomotor y las alteraciones genitourinarias (tabla 2).
Adicionalmente, otras características presentes fueron la voz aguda y las alteraciones dentales, las comisuras labiales descendentes, la micrognatia, la hiperhidrosis, la hipotonía muscular y el cierre tardío de fontanelas, todas ellas en mayor proporción que la previamente reportada. En proporción menor a lo reportado se encontraron las anomalías de pabellones auriculares (tabla 2).
Aun cuando esta serie de pacientes fue pequeña, se pudo corroborar que al utilizar los criterios clínicos propuestos por Saal6 es posible tener una mejor aproximación al diagnóstico clínico de SSR. Cuando se consideraron los criterios de Dias y colaboradores25, 8/9 pacientes presentaron tres de las cuatro características diagnósticas mientras que el paciente restante presentó todas. Con el resto de los criterios propuestos, incluidos los de Bartholdi y colaboradores24 que son los que utilizan mayor número de características clínicas, también se confirmó el diagnóstico de SSR en todos los casos. Los 11 casos en los que se excluyó el diagnóstico utilizando los criterios de Saal6 tampoco cumplieron los criterios diagnósticos de las otras escalas.
El diagnóstico del SSR es fundamentalmente clínico ya que, si bien se conocen varias causas moleculares, hasta en el 50% de los pacientes con diagnóstico clínico de certeza no se identifica su etiología. En los casos en los que se determina la alteración molecular del SSR es posible realizar una relación con las características clínicas presentes y brindar un asesoramiento genético con bases moleculares. Así, se ha reportado que en los casos con epimutación en 11p15.5 el cuadro clínico presenta con mayor frecuencia asimetría corporal y frontal prominente en comparación con aquellos con DUP(7)mat u otras causas1,4,5,7,26. Por el contrario, las alteraciones faciales como la cara triangular, el frontal prominente, micrognatia, anomalías dentales y de pabellones auriculares, la hipotonía muscular y el retraso en el desarrollo psicomotor prevalecen en los pacientes con DUP(7)mat1,4,5,7,26. La causa más frecuentemente identificada en SSR son las epimutaciones; en esta serie solo los pacientes 4, 5 y 9 presentaron asimetría corporal (tabla 2), por lo que podrían corresponder a esta etiología. Sin embargo, se debe considerar que si la aplicación de los criterios clínicos diagnósticos es, por demás, estricta podría correrse el riesgo de dejar fuera a pacientes que muestran un fenotipo más sutil, como lo han sugerido Eggermann y colaboradores7. Con relación al asesoramiento genético, ante un diagnóstico clínico de SSR debido a un defecto en la impronta en CRI1, el riesgo de recurrencia para otros miembros de la familia raramente se encuentra aumentado respecto a la población general. Cuando la etiología es por DUP(7)mat el riesgo es el mismo que el de la población general6. Sin embargo, cuando se trata de una duplicación o mutación del CRI2 en el cromosoma 11p15.5 materno, el riesgo de recurrencia es del 50% cuando lo transmite la madre35.
En este reporte de pacientes con SSR, que recoge la experiencia en uno de los centros de referencia pediátricos nacionales en el periodo comprendido entre 1998 y 2012, se refleja la complejidad diagnóstica del SSR basada únicamente en criterios clínicos. No existe hasta la fecha un índice internacionalmente aceptado para su diagnóstico; sin embargo, una opción de amplia consideración son los criterios propuestos por Saal6 que fueron aplicados en los pacientes aquí estudiados. Este análisis demostró que cuando se aplican criterios estrictos en solo el 45% de los pacientes en quienes se consideró inicialmente el diagnóstico de SSR correspondió a esta entidad. Este hecho refleja la importancia que tiene para el médico general y el pediatra llevar a cabo una valoración estrecha del crecimiento y desarrollo de todos los pacientes en las diferentes etapas, y ser muy cuidadosos en la evaluación de las dismorfias pequeñas o de los criterios de apoyo diagnóstico, en particular en pacientes en estudio por talla baja. Este abordaje permitirá descartar otras causas e identificar a aquellos pacientes que podrían beneficiarse de estudios moleculares para descartar entidades sindrómicas, como el SSR, lo que tendrá implicaciones importantes para el manejo, pronóstico y asesoramiento genético.
FinanciaciónProtocolo de investigación «Implicaciones del patrón de metilación en la región 11p15.5 como mecanismo etiológico de la hemihiperplasia aislada» (HIM/2012/007), Fondos Federales.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.