



La sarcoidosis es una enfermedad sistémica de etiología desconocida que raramente se presenta en la infancia. Generalmente afecta los pulmones; sin embargo, puede involucrar diversos órganos. Ocasionalmente afecta el estado general, y origina fiebre, hepatomegalia y esplenomegalia.
Caso clínicoSe presenta el caso de un adolescente de doce años de edad con sarcoidosis infantil de inicio tardío, cuyo diagnóstico fue confirmado con un estudio histopatológico de ganglio linfático. El paciente cursó con afección general, hipercalcemia, eritema nodoso, alteraciones pulmonares graves, adenopatías, hepatomegalia y masa testicular. Recibió tratamiento con esteroides, con excelente respuesta clínica.
ConclusionesSe resalta la importancia de considerar el diagnóstico de sarcoidosis en los pacientes con hepatomegalia, adenopatías, daño pulmonar difuso, eritema nodoso, masa testicular e hipercalcemia, así como la necesidad del abordaje multidisciplinario para valorar el compromiso orgánico múltiple y el inicio oportuno de la terapia con esteroides, con el fin de evitar la progresión de la enfermedad.
Sarcoidosis is a systemic disease of unknown etiology that rarely occurs in children. It usually affects the lungs, however, it may involve various organs. It occasionally affects the general condition, and causes fever, hepatomegaly and splenomegaly.
Case reportWe report the case of a twelve-year-old adolescent with late-onset childhood sarcoidosis which diagnosis was confirmed by lymph node histopathological study. The patient presented general condition, hypercalcemia, erythema nodosum, severe lung disorders, lymphadenopathy, hepatomegaly and testicular mass. He received treatment with steroids, with excellent clinical response.
ConclusionsWe highlight the importance of considering the diagnosis of sarcoidosis in patients with hepatomegaly, lymphadenopathy, diffuse lung damage, erythema nodosum, testicular mass and hypercalcemia, as well as the need for a multidisciplinary approach to assess multiple organ involvement and the early beginning of steroid treatment in order to prevent the progression of the disease.
La sarcoidosis es una enfermedad sistémica crónica de etiología desconocida y de distribución mundial, que afecta principalmente a la población adulta. Su presentación en la infancia resulta muy rara.
La enfermedad afecta principalmente a los pulmones; sin embargo, puede involucrar otros órganos como ojos, piel, ganglios linfáticos y articulaciones. Con menos frecuencia compromete al sistema nervioso, corazón y aparato genitourinario, causando nefrolitiasis y masa testicular; en algunos casos se presenta como fiebre de origen desconocido con hepatomegalia y esplenomegalia.
El diagnóstico de sarcoidosis se realiza por exclusión. Por ello, una vez que se tiene la sospecha clínica, una biopsia de los órganos involucrados mostrará, como hallazgo histopatológico característico, la presencia de granulomas epitelioides no caseificantes1.
Las primeras descripciones sobre la sarcoidosis fueron realizadas en Europa a finales del siglo XIX. En 1877, en Inglaterra, destacaron los estudios de J. Hutchinson en un paciente con lesiones crónicas en la piel, artritis e insuficiencia renal crónica, quien dio a las alteraciones cutáneas el término de psoriasis papilar. En Francia, en 1889, E. Besnier describió también las lesiones de la piel con el nombre de lupus pernio. Por otro lado, en 1899 en Dinamarca, C. Boeck designó a las lesiones histológicas de la piel con el término sarkoid, por su similitud con el sarcoma. En 1914, en Suecia, J. N. Schaumann describió la forma sistémica de la enfermedad; además, señaló que tanto el lupus pernio de Besnier como el sarkoid de Boeck eran manifestaciones de una misma enfermedad, ya que los tejidos afectados en estos pacientes mostraban la formación de granulomas, a los que llamó linfogranulomatosis benigna, con el fin de diferenciarlos del granuloma maligno de Hodgkin. El compromiso neurológico en la sarcoidosis fue reportado por C. Heerfordt en 1923, quien describió pacientes con fiebre uveo-parotídea y compromiso de los nervios craneanos.
La forma pulmonar aguda con adenopatías mediastinales acompañada de eritema nodoso, artritis y uveitis fue descrita en Suecia en el año 1953 por S. Löfgren.
Durante más de 130 años la mayor parte de los estudios sobre sarcoidosis se han realizado en adultos. Sin embargo, desde 1923 se han descrito casos en niños. Al padecimiento se le conoció como enfermedad de Besnier-Boeck-Schaumann hasta 1958, cuando se realizó el Congreso Mundial de Sarcoidosis en Londres y se generalizó el término sarcoidosis para designar a la enfermedad2,3.
En el momento actual, de acuerdo con el consenso internacional establecido por la American Thoracic Society, la European Respiratory Society y la World Association for Sarcoidosis and Other Granulomatosis, se considera que la sarcoidosis es una enfermedad sistémica granulomatosa, cuya etiología continúa siendo desconocida, que por lo general afecta a los adultos, y es muy rara en los niños4,5.
La incidencia y la gravedad de la sarcoidosis varían en distintas regiones del mundo, así como en diferentes grupos étnicos, probablemente debido a variaciones en las exposiciones ambientales, la prevalencia de alelos HLA y otros factores genéticos. Los países escandinavos, Inglaterra, Estados Unidos y Japón presentan la más alta prevalencia de la enfermedad; en Suecia, la tasa de morbilidad en la población general es de 64/100,000 y en los Estados Unidos, de 35/100,000 habitantes.
Existen muy pocos datos epidemiológicos en la población infantil. Se estima que la tasa de morbilidad de la sarcoidosis en la infancia es de 0.29/100,000; sin embargo, varía desde 0.06/100,000 en menores de 4 años hasta 1.02/100,000 en adolescentes de 14 a 15 años de edad6–8.
En cuanto a la mortalidad por sarcoidosis, durante el periodo 1999-2010, el National Center for Health Statistics reportó un total de 29,176,040 defunciones, de las cuales en 10,348 casos la causa de la muerte fue la sarcoidosis, obteniendo una tasa promedio de 2.8/1,000,000 habitantes. Sin embargo, la mortalidad en la población afro-americana fue 12 veces mayor que en la población caucásica9.
En México se ha considerado que la incidencia de sarcoidosis es baja, probablemente debido a factores genéticos o al subregistro de casos. Por lo tanto, no se cuenta con estudios sobre la epidemiología de la sarcoidosis infantil, ya que en los últimos 20 años solamente se han reportado trece casos, donde destacan el caso de un adolescente con afección pulmonar, publicado por el Instituto Nacional de Enfermedades Respiratorias, y otro estudio realizado en el Instituto Nacional de Pediatría sobre dos casos de sarcoidosis de inicio temprano con manifestaciones cutáneas10–12.
La sarcoidosis es una enfermedad inflamatoria crónica que resulta de la acción de un agente ambiental que desencadena una respuesta inmune inicial de células T helper tipo 1 (Th1), y lleva al desarrollo de granulomas no caseificantes con afectación sistémica en individuos genéticamente susceptibles.
De acuerdo con los hallazgos epidemiológicos del estudio multicéntrico ACCESS (A Case Control Etiologic Study of Sarcoidosis), hay un aumento del riesgo para padecer sarcoidosis con algunas condiciones ambientales que generan estímulos antigénicos, los cuales actúan como agentes desencadenantes del proceso. Destacan diversos factores como materiales orgánicos (polen de plantas, hojas de pino y semillas), materiales inorgánicos (sílice, berilio, circonio, titanio, aluminio y fibra de vidrio) o microorganismos (Mycobacterium tuberculosis, Propionibacterium acnes, Brucella, Borrelia, Leptospira, Mycoplasma, Leishmania y Schistosoma). Sin embargo, aunque no se ha identificado de manera concluyente algún agente infeccioso (con cultivos o marcadores de RNA ribosómico en tejidos obtenidos de biopsias), la hipótesis de un agente microbiológico es la más aceptada ya que existe evidencia clínica y epidemiológica de la transmisibilidad de la sarcoidosis. Esto se ha observado en pacientes receptores de trasplantes que han desarrollado la enfermedad después del trasplante de tejidos u órganos procedentes de donadores con sarcoidosis. Así mismo, se ha reportado el desarrollo de sarcoidosis en el pulmón trasplantado en pacientes que padecían la enfermedad; del mismo modo, se ha logrado inducir experimentalmente la formación de granulomas en animales implantados con tejido afectado de pacientes con sarcoidosis13–18.
La existencia de un factor genético predisponente explica la incidencia más alta del padecimiento en los familiares de enfermos con sarcoidosis, así como las diferencias en la prevalencia y el curso clínico de la enfermedad en distintos grupos étnicos.
Los estudios recientes con técnicas de biología molecular han demostrado que las alteraciones genéticas relacionadas con alto riesgo para sarcoidosis se ubican en el complejo mayor de histocompatibilidad (MHC), localizado en el brazo corto del cromosoma 6, en los antígenos de histocompatibilidad HLA de clase I, como el HLA-B7 y HLA-B8, y sobre todo en los alelos de HLA de clase II, como HLA-DR5, HLA-DR6, HLA-DR8 y HLA-DR9, relacionados con alto riesgo en Asia, y el HLA-DRB1 en la población afroamericana. Por otra parte, en la población europea se encontraron HLA-DR14 y HLA-DR15, relacionados con la enfermedad crónica, el HLA-DR3 con la forma aguda, y el HLA-DR17 con las formas autolimitadas de la sarcoidosis.
Los mecanismos inmunes que causan la sarcoidosis no se conocen por completo, pero se considera que los macrófagos no pueden realizar adecuadamente el reconocimiento y presentación del antígeno a los linfocitos T. Sin embargo, en los enfermos de sarcoidosis no existe evidencia de deficiencias en la inmunidad celular o humoral.
El proceso comienza cuando el antígeno que ingresó al macrófago y a la célula dendrítica es presentado en el sitio MHC. Esto permite al linfocito T localizar y unirse al complejo MHC-II, lo que lleva a su activación y expansión clonal, demostrada con el incremento en la transcripción del RNAm del receptor de la superficie celular de la célula T (TCR), y con la presencia de marcadores de activación antígeno-específica de linfocitos T, como el CD69, la glucoproteína 240 y el antígeno muy tardío-1 (VLA-1).
Después ocurre la diferenciación de linfocitos T-CD4 al fenotipo de linfocitos T-helper 1 (Th1), con liberación de citocinas, como interferón-γ (IFN-γ), factor de necrosis tumoral-α (TNF-α), factor de crecimiento transformante-β (TGF-β), IL-1β, IL-2, IL-12, IL-15 e IL-18, además de varias quimosinas, como la proteína inflamatoria del macrófago 3α (MIP3α), proteína interferón inducible 10 (IP-10), proteína quimiotáctica del monocito (MCP-1) y la CCL5 o RANTES (Regulated on Activation, Normal T-cell Expressed and Selected), que por su efecto quimiotáctico mantienen el reclutamiento de células en los sitios de formación de granulomas.
Se ha observado que en los pacientes con sarcoidosis crónica activa se presenta una alteración en la función de las células T reguladoras, lo cual permite la persistencia del proceso inflamatorio con progresión de los granulomas. Por el contrario, en enfermos con síndrome de Löfgren, que es una forma de sarcoidosis aguda con una tasa elevada de remisión, se ha encontrando un incremento de las células T reguladoras que disminuyen la proliferación de linfocitos y la producción de citocinas.
En algunos pacientes, la inflamación inicial de tipo Th1 es sustituida por una respuesta con predominio de células T-helper 2 (Th2), caracterizada por una disminución del IFN-γ, persistencia de la actividad del TNF-α y del TGF-β, producción de la enzima convertidora de angiotensina (ECA), neopterina e IL-8, lo cual reduce la formación de granulomas; sin embargo, favorece el desarrollo de fibrosis debido a la actividad de algunas citocinas de los macrófagos, como el factor de crecimiento insulínico tipo 1 (IGF-1), el factor de crecimiento plaquetario, la producción de IL-4 e IL-13, que generan activación de los fibroblastos con depósito de fibronectina y colágena en el espacio extracelular. La causa de la progresión a esta forma fibro-proliferativa no es clara, pero puede implicar la pérdida de los mecanismos apoptóticos, la pérdida de la respuesta reguladora, o la persistencia de un antígeno que no puede ser reconocido ni procesado adecuadamente, lo cual ocasiona cicatrización y reparación fibrosa del tejido afectado con daño crónico e irreversible19–24.
Se ha demostrado que la deficiencia de vitamina D conlleva un mayor riesgo de desarrollar sarcoidosis, debido a que las células presentadoras de antígeno (macrófagos, monocitos y células dendríticas) tienen el receptor de la vitamina D. Esta vitamina inhibe la activación de los macrófagos inducida por el IFN-γ y disminuye la actividad presentadora del antígeno MHC de clase II de los macrófagos; por lo tanto, se considera que esta deficiencia conduce a una regulación alterada de los linfocitos Th1, permitiendo una respuesta inmune persistente25–27.
Además, se ha comprobado que los macrófagos activados son capaces de producir vitamina D3 activa (1,25-hidroxicolecalciferol o calcitriol) y el péptido relacionado con paratohormona (PTHrP), que podrían contribuir a la hipercalcemia e hipercalciuria frecuentemente presentes en la enfermedad28,29.
La paradoja inmunológica es uno de los hallazgos más notables en la sarcoidosis. Esta paradoja está caracterizada por una intensa respuesta inmune celular en los órganos afectados en contraste con una situación de anergia periférica, que se manifiesta como la falta de respuesta al PPD y a otras pruebas intradérmicas. La posible explicación para este fenómeno se debe a la presencia de linfopenia en sangre periférica, y sobre todo a la actividad de los linfocitos T CD8+ supresores.
Otro fenómeno que también ha sido objeto de interés y controversia es la formación de granulomas en la piel de enfermos con sarcoidosis, de 4 a 6 semanas después de la aplicación del extracto de tejido sarcoideo en la prueba de Kveim-Siltzbach (utilizada anteriormente con fines diagnósticos)30,31.
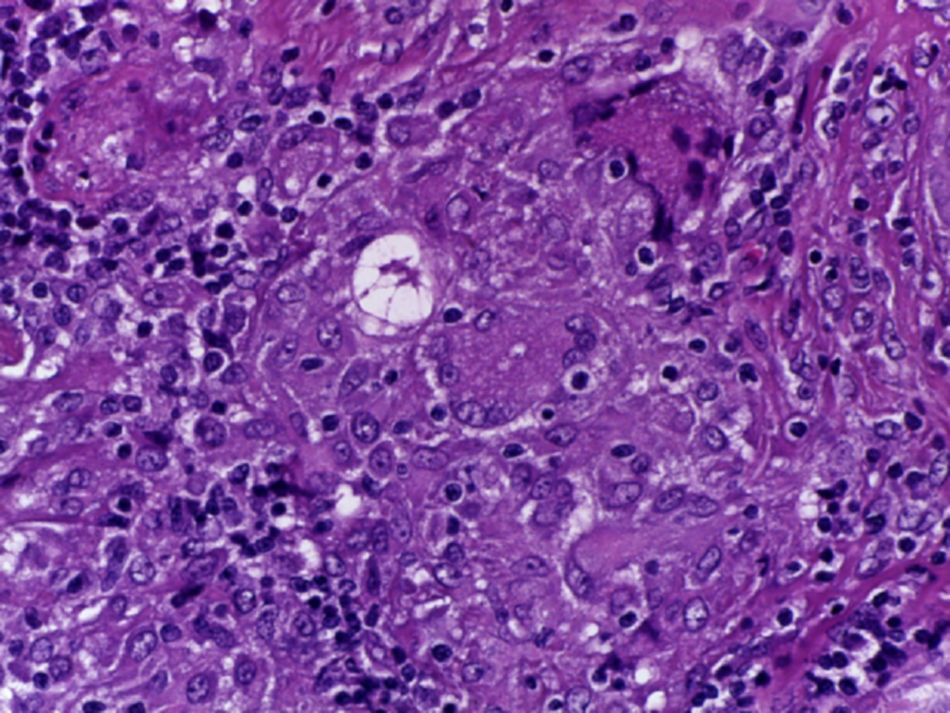
El hallazgo histológico característico de la sarcoidosis es la presencia de granulomas epitelioides no caseificantes esparcidos de manera difusa en diferentes tejidos, pero que principalmente afectan al ganglio linfático.
Los granulomas de la sarcoidosis tienen aspecto compacto, con límites bien definidos. Pueden encontrarse en distintas etapas de evolución, que van desde granulomas altamente celulares hasta lesiones con celularidad disminuida, con fibrosis o con hialinización progresiva; además, no presentan necrosis central ni evidencia de cuerpos extraños, lo cual permite diferenciarlos de los granulomas observados en infecciones por micobacterias, micosis sistémicas e infestaciones por metazoarios.
El granuloma típico de la sarcoidosis presenta dos zonas características que se describen a continuación:
- 1.
La zona central o folículo, constituida por un denso conglomerado de células epitelioides en forma de racimos, acompañados de linfocitos, macrófagos, células gigantes multinucleadas de Langhans, mastocitos y fibroblastos. En el 60% de los casos se observan estructuras de aspecto estelar (o cuerpos asteroides) y estructuras laminares llamadas cuerpos de Schaumann, de 1-15μm de diámetro y PAS+. En estudios con métodos inmunohistoquímicos, la zona central de un granuloma activo consiste en macrófagos en diversas etapas de activación y diferenciación. Esta zona está rodeada por células T CD4+ intercaladas con un pequeño número de células T CD8+ y células B.
- 2.
La zona periférica, formada por un anillo de linfocitos, monocitos y fibroblastos. En la periferia del granuloma se acumulan las células T reguladoras CD3+/CD4+/CD25+/ +Fox P3, además de células T CD8+ y fibroblastos, que cuando disminuye la actividad de los granulomas, producen la fibrosis.
En la forma crónica de la enfermedad, los granulomas pueden estar encapsulados por un halo fibroso o pueden ser sustituidos por cicatrices de tejido fibroso y hialino32–34.
La presentación clínica de la sarcoidosis en los niños es muy variada, ya que depende del grado de extensión de la enfermedad y de los órganos involucrados. En la mayoría de los casos de sarcoidosis infantil se produce afección a múltiples órganos, con respuesta inflamatoria difusa, que genera síntomas generales como fiebre, astenia, hiporexia, náuseas y pérdida de peso, además de los síntomas y signos específicos derivados de la disfunción de cada órgano afectado.
La sarcoidosis en la infancia puede presentarse en dos formas clínicas:
- 1.
Sarcoidosis de inicio temprano o síndrome de Blau. Se presenta antes de los cinco años de edad en forma esporádica o familiar. Se asocia con mutaciones del gen NOD2/CARD15 localizado en el cromosoma 16. En el 75% de estos casos se manifiesta con una triada caracterizada por poliartritis, uveítis y erupción cutánea, y en el 25% restante se acompaña de afección a otros órganos.
- 2.
Sarcoidosis de inicio tardío. Se desarrolla en los niños mayores de 5 años y se asemeja a la forma clínica del adulto. No está asociada con mutaciones del gen NOD2/CARD15; se caracteriza por la presencia de fiebre, afectación multiorgánica, sobre todo pulmonar, cutánea, neurológica, ocular, renal, articular, adenopatías, hepatomegalia y esplenomegalia35,36.
El pulmón se encuentra comprometido en más del 90% de los casos; sin embargo, el espectro clínico de presentación es muy variado. La mayoría de los casos se presentan en forma aguda, con ataque al estado general y síntomas respiratorios inespecíficos como tos seca, disnea e hiperreactividad bronquial, pero también la enfermedad puede iniciar en forma insidiosa y con mínima sintomatología. La afección se localiza principalmente en el intersticio pulmonar, con aumento de tamaño de los ganglios linfáticos hiliares, traqueo-bronquiales y mediastinales.
Aproximadamente el 70% de los casos remiten espontáneamente, pero el 30% restante evoluciona a la cronicidad, donde la enfermedad provoca cambios irreversibles en el parénquima pulmonar, con fibrosis y formación de neumatoceles que conducen a la insuficiencia respiratoria crónica y a la muerte.
Las pruebas de funcionamiento respiratorio, como la espirometría y la pletismografía, muestran alteración con un patrón de tipo restrictivo, y son de gran utilidad para valorar la evolución del paciente y la respuesta al tratamiento.
Las anomalías radiológicas encontradas en sarcoidosis pulmonar se han estatificado en cuatro estadios radiográficos, que permiten elegir el plan terapéutico y establecer un pronóstico según el estadio encontrado en el momento del diagnóstico. Adicionalmente, el comportamiento en los dos primeros años de la enfermedad, de tal manera que la posibilidad de remisión de las lesiones disminuye de acuerdo con el grado de evolución del padecimiento: mientras la resolución radiográfica en los primeros tres estadios de la enfermedad es factible, durante en el estadio IV, que se considera la etapa terminal, las lesiones radiográficas son ya irreversibles37 (tabla 1).
Estadificación radiográfica de la sarcoidosis torácica*
| Estadio | Hallazgos | Remisión |
|---|---|---|
| Estadio 0 | Radiografía de tórax normal | > 90% |
| Estadio I | Adenopatías hiliares bilaterales sin compromiso pulmonar | 60-90% |
| Estadio II | Adenopatías hiliares bilaterales más infiltrados pulmonares | 40-60% |
| Estadio III | Infiltrados pulmonares sin adenopatías hiliares | 10-20% |
| Estadio IV | Fibrosis extensa con distorsión y bulas enfisematosas; pulmón en panal | 0% |
La incidencia de afección ocular varía del 30 al 70%. La lesión característica es la uveítis. Sin embargo, puede afectarse la conjuntiva, esclerótica, cristalino y glándulas lacrimales, produciendo cataratas, glaucoma y querato-conjuntivitis seca. En el segmento posterior puede ocurrir vitritis, vasculitis retiniana isquémica con neovascularización, oclusión de la vena central de la retina, afección del nervio óptico, edema macular, lesiones perforantes, exudados en “gotas de cera” y desprendimiento de retina con ceguera, las cuales pueden detectarse en el examen oftalmológico con lámpara de hendidura y con angiografía retiniana38,39.
Las alteraciones cutáneas se presentan en el 20 al 30% de los pacientes. La lesión más frecuente es el eritema nodoso, que se utiliza como marcador de sarcoidosis aguda y que generalmente remite en 6-8 semanas. Cuando se acompaña de fiebre, artralgias y adenopatías hiliares, se constituye el síndrome de Löfgren. Otras lesiones cutáneas incluyen nódulos subcutáneos, placas similares a psoriasis, alopecia, lesiones hiperpigmentadas y vasculitis leucocitoclástica.
Las afecciones articulares se presentan hasta en el 80% de los casos. Pueden ser en forma de poliartritis migratoria y/o artritis persistente, además de tenosinovitis granulomatosa. Cuando se presenta en menores de cinco años con poliartritis, exantema y uveítis, constituye el síndrome de Blau40.
El compromiso del sistema nervioso se presenta en el 10% de los pacientes, por lo general con afección de los pares craneales II y VII como resultado de una meningitis granulomatosa. Sin embargo, en estudios realizados en autopsias se encontró compromiso del SNC hasta en el 25% de los casos de defunción por sarcoidosis.
La afectación cardiaca se diagnostica clínicamente en menos del 5% de los pacientes con sarcoidosis, aunque se ha encontrado que en el 20 al 30% de los estudios post mórtem existe compromiso del sistema de conducción, que puede ocasionar arritmias y muerte súbita. Los hallazgos ecocardiográficos consisten en disfunción ventricular y disminución de la fracción de eyección del ventrículo izquierdo; sin embargo, la comprobación histológica requiere de biopsias miocárdicas por cateterismo cardiaco.
La afección de las glándulas exocrinas es un hallazgo frecuente. Puede ocurrir afección parotídea y de las glándulas salivales menores hasta en el 60% de los casos, y con menos frecuencia se ha reportado pancreatitis granulomatosa por sarcoidosis.
La afectación renal se presenta en el 10% de los pacientes debido a hipercalcemia y nefrocalcinosis, además de litiasis renal.
El compromiso genitourinario se ha reportado en el 0.2% de los casos diagnosticados clínicamente, y en el 5% de estudios de autopsias, siendo la alteración más frecuente la masa escrotal por afección del testículo y del epidídimo41–48.
El diagnóstico de la sarcoidosis infantil se realiza con los mismos criterios establecidos para los adultos, publicados en el consenso de la American Thoracic Society y de la European Respiratory Society en 1999, los cuales consisten en lo siguiente:
- 1.
Cuadro clínico compatible.
- 2.
Evidencia histológica de granulomas no caseificantes en las biopsias obtenidas de los órganos afectados.
- 3.
Exclusión de otros procesos patológicos que pueden manifestarse con un cuadro clínico e histopatológico similar, especialmente las infecciones por micobacterias y hongos, así como otros procesos inmunológicos.
No existe disponible una prueba definitiva que confirme el diagnóstico de sarcoidosis. Sin embargo, los estudios que ayudan a comprobar la enfermedad son los que a continuación se mencionan:
- 1.
El análisis citológico del lavado bronco alveolar. Es una prueba de gran utilidad ya que en los enfermos con sarcoidosis se encuentra una celularidad predominante de linfocitos en >90% de los casos. Además, el estudio con citometría de flujo revela una relación CD4/CD8 > 3.5, la cual tiene una sensibilidad del 52-59% y una especificidad del 94-96% para el diagnóstico de sarcoidosis49–51.
- 2.
La prueba de Kveim-Siltzbach. Se ha utilizado para el diagnóstico de la sarcoidosis desde 1941. Consiste en la inoculación intradérmica de una suspensión obtenida de tejido esplénico de pacientes con sarcoidosis; 6 semanas después debe realizarse una biopsia de piel en el sitio inoculado para la búsqueda de granulomas. La prueba tiene una sensibilidad del 75% y una especificidad > 90%, pero en los últimos años su aplicación clínica ha sido muy restringida debido a la dificultad para obtener el reactivo; además, se requiere que los pacientes no hayan recibido esteroides, e incluso algunos autores la han desaconsejado por el riesgo de transmisión de enfermedades infecciosas51,52.
- 3.
La determinación de los niveles séricos de la enzima convertidora de angiotensina (ECA). La ECA ha sido utilizada en el diagnóstico de la enfermedad, debido a que el 80% de los enfermos con sarcoidosis infantil presentan elevación de la misma. Además, es de gran utilidad como un marcador de actividad de la enfermedad ya que los niveles de la enzima descienden en los pacientes que entran en remisión.
- 4.
La medición del receptor soluble de la interleucina-2 (sIL-2R). Es una prueba que ha sido muy útil en la evaluación de la actividad de la sarcoidosis53–55.
- 5.
La determinación de los niveles de calcio sérico y urinario. Son de gran utilidad tanto en el diagnóstico como en el monitoreo de la evolución de la enfermedad, ya que la hipercalcemia que se presenta en el 20% de los casos y la hipercalciuria hasta en el 60% de los enfermos con sarcoidosis dependen de la actividad y la extensión de la enfermedad56–60.
Paciente de sexo masculino de 12 años de edad con ambos padres y cuatro hermanos sanos, sin otros antecedentes familiares relevantes. Fue el producto de una primera gestación de término, obtenido por parto eutócico sin complicaciones perinatales, desarrollo psicomotor adecuado y esquema de vacunación completo. No se refirieron antecedentes personales patológicos, excepto que presentó varicela a los 6 años de edad, sin complicaciones.
Su padecimiento se caracterizó por tos seca en accesos aislados, de un año de evolución, pérdida de peso de 12kg en 6 meses, astenia, hiporexia, palidez, náusea y vómitos ocasionales de contenido gástrico. Presentó aumento de volumen en región supraclavicular izquierda y nódulos subcutáneos indoloros en ambos antebrazos, de 4 meses de evolución. Una semana antes de su ingreso acudió al hospital de su localidad por dolor tipo cólico en flanco derecho, con signo de Giordano ipsilateral. El ultrasonido abdominal mostró hepatomegalia y litiasis renal derecha, por lo cual fue referido a esta institución.
Al examen físico se encontró un adolescente del sexo masculino consciente y bien orientado, caquéctico, pálido, ojos con pupilas isocóricas con reflejo fotomotor adecuado, oídos, nariz y faringe normales. Pares craneales íntegros. Cuello sin ingurgitación yugular, ganglios cervicales de 0.5 a 1cm y un ganglio supraclavicular izquierdo de 2cm de diámetro, móviles, indoloros, sin eritema ni aumento de temperatura. Tórax adelgazado con taquipnea y discreta retracción intercostal, movimientos de amplexión y amplexación normales, murmullo vesicular adecuado sin estertores ni sibilancias, ruidos cardiacos rítmicos sin soplos. Abdomen blando, hepatomegalia de 6cm y esplenomegalia de 4cm, ruidos peristálticos normales. Genitales masculinos Tanner II, escroto asimétrico por aumento de volumen indoloro del testículo izquierdo y transiluminación negativa. Extremidades hipotróficas, con eritema nodoso localizado en antebrazos y piernas, pulsos periféricos simétricos, reflejos osteo-tendinosos normales sin signos de Babinski ni sucedáneos.
Inicialmente se manejó con soluciones parenterales, analgésico y aporte de oxígeno por mascarilla facial. Al mejorar la taquipnea, se inició la alimentación por vía oral con adecuada tolerancia y el abordaje diagnóstico.
Los exámenes de laboratorio iniciales mostraron los siguientes resultados: hemoglobina 14.4g/dl, leucocitos 7,600/mm3, neutrófilos 48%, bandas 4%, linfocitos 37%, monocitos 7% plaquetas 286,000/mm3, velocidad de sedimentación globular 38mm/h. Proteína C reactiva<0.319. La química sanguínea mostró glucosa 91mg/dl, creatinina 1.5mg/dl, BUN 17mg/dl, ácido úrico 6mg/dl, proteínas séricas 8.6g/dl, albúmina 3.2g/dl, globulinas 5.4g/dl, bilirrubina total 0.36mg/dl, bilirrubina directa 0.9mg/dl, bilirrubina indirecta 0.27mg/dl, ALT 35 U, AST 26 U, colesterol 120mg/dl, triglicéridos 160mg/dl, Na 129 mmol/l, K 3.6 mmol/l, Cl 98 mmol/l, Ca 13mg/dl, P 4.2mg/dl, Mg 2.4mg/dl, osmolaridad sérica 268 mOsm/l. Gasometría arterial sin aporte de oxígeno: pH 7.43, pCO2 32.4mmHg, pO2 49.9mmHg, HCO3 21.3 mmol/l, sO2 83%, lactato 2.4 mmol/l, anión Gap 13.6 mmol/l. TP 12.4”, TTP 29.3”.
Los estudios serológicos para virus de hepatitis A, B y C, CMV, VEB, varicela zoster, virus herpes simple tipo I y II, negativos. ELISA para VIH, negativa. Los anticuerpos Anti-DNA ds, anticuerpos antinucleares (ANA), anticuerpos anticitoplasma de neutrófilos (p-ANCA y c-ANCA) y VDRL, negativos.
El examen general de orina resultó con aspecto turbio, densidad 1.020, pH 6.0, eritrocitos abundantes, leucocitos 1-2/campo, nitritos negativos, proteínas 75mg/dl, sedimento con cristales de oxalato de calcio y de fosfato amorfo.
Calciuria en orina de 24 horas de 10.9mg/kg/día. Relación calcio/creatinina (Ca/cr) en micción aislada de 0.52mg/mg. Fosfaturia en orina de 24 h de 63.1mg/kg/día. Reabsorción tubular de fosfatos del 63.3%. Relación fósforo/creatinina (P/cr) en micción aislada de 0.80mg/mg. Depuración de creatinina de 70.4mg/ml/min.
Cistatina C sérica, 2.18mg/l (0.53-0.95mg/l). Ácido oxálico en orina de 24h, 53.7mg (< 38mg). Acido cítrico en orina de 24h, 427mg (100-1300mg).
Hormona paratiroidea (PTH)<1.20 pg/ml (10-55 pg/ml). Calcitriol (1,25 dihidroxi-vitamina D3), 30.7 pg/ml (19.9-79.3 pg/ml). Enzima convertidora de angiotensina (ECA), 272 U/l (13-100 U/l). Alfa-fetoproteína, 1.07 ng/ml (< 10 ng/ml). Beta-gonadotropina, 1.20 mIU/ml (< 5 mIU/ml).
Prueba de PPD con testigo de candidina, negativa. Búsqueda de BAAR en tres muestras de jugo gástrico, negativas.
El aspirado de médula ósea mostró celularidad disminuida, megacariocitos disminuidos, metamielocitos 2.5%, bandas 10.5%, segmentados 35%, eosinófilos 3%, linfocitos 23.5%, blastos 1.5% y normoblastos 24%.
Inmunoglobulinas séricas: IgA 724mg/dl (108-325mg/dl), IgE 82.6mg/dl (0-100mg/dl), IgM 175mg/dl (70-150mg/dl), IgG 2710mg/dl (770-1510mg/dl).
Complemento C3 113mg/dl (90-180mg/dl), C4 16.6mg/dl (10-40mg/dl).
Pruebas de reducción de nitroazul de tetrazolio y quimioluminiscencia en reposo y con estímulo, normales.
Receptor soluble de Interleucina-2 (sIL-2R), 3230 U/ml (406 -1100 U/ml).
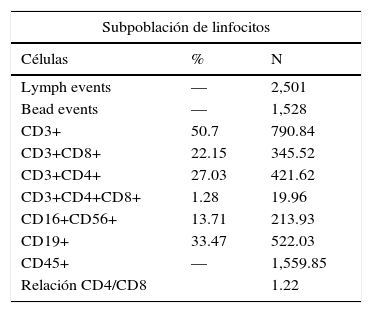
La citometría de flujo en sangre periférica mostró la población de linfocitos T y B cuantitativamente normal, con una relación CD4/CD8=1.22 (tabla 2).
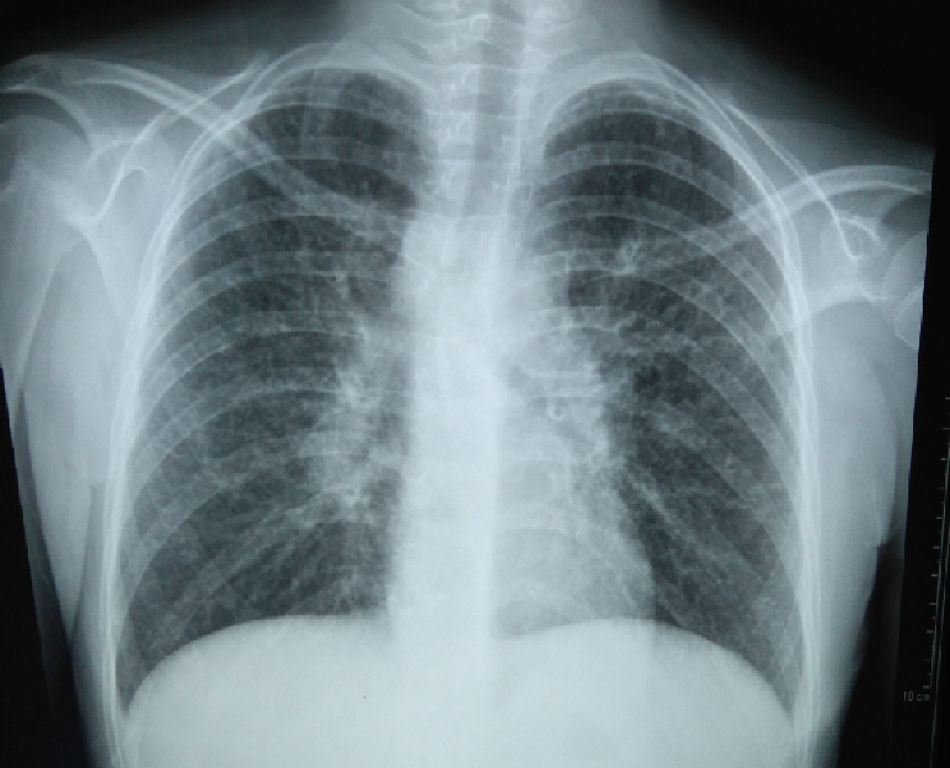
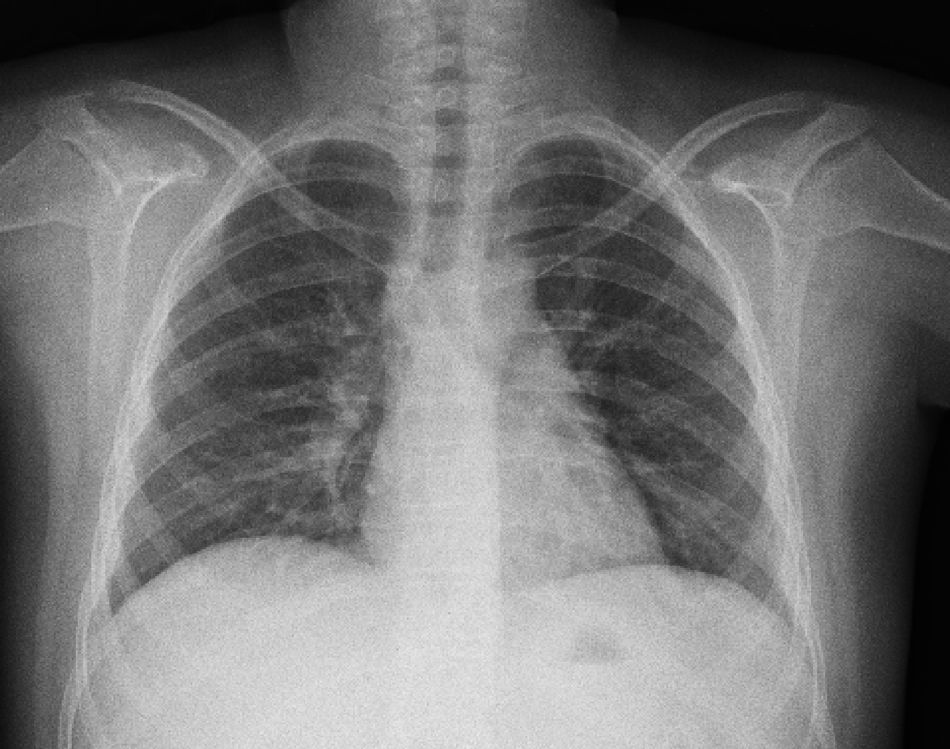
La radiografía de tórax inicial mostró infiltrado micronodular y reticular difuso con adenopatías hiliares bilaterales (fig. 1).

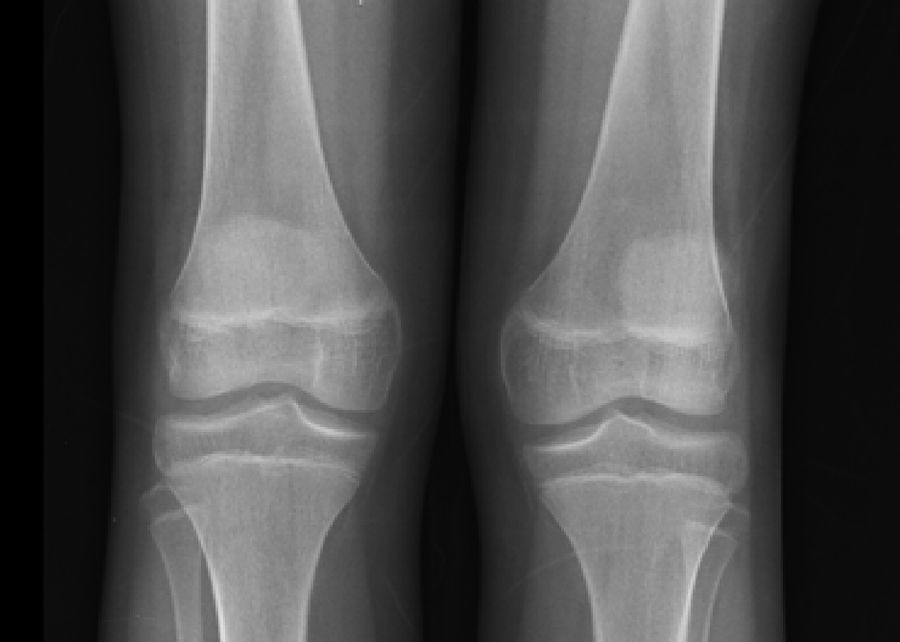
Las radiografías de cráneo se observaron normales, aunque los estudios de huesos largos con evidencia de descalcificación (fig. 2).

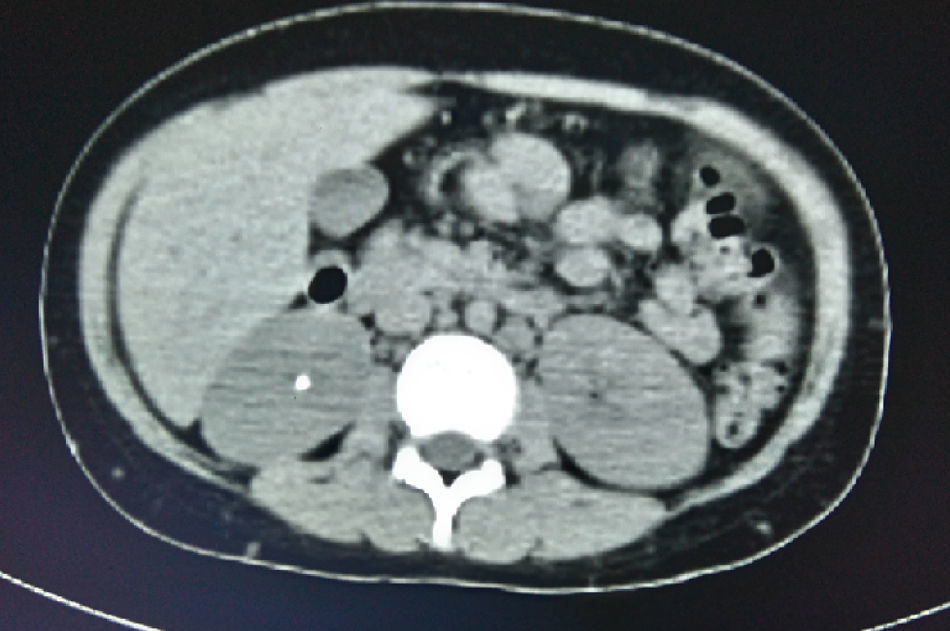
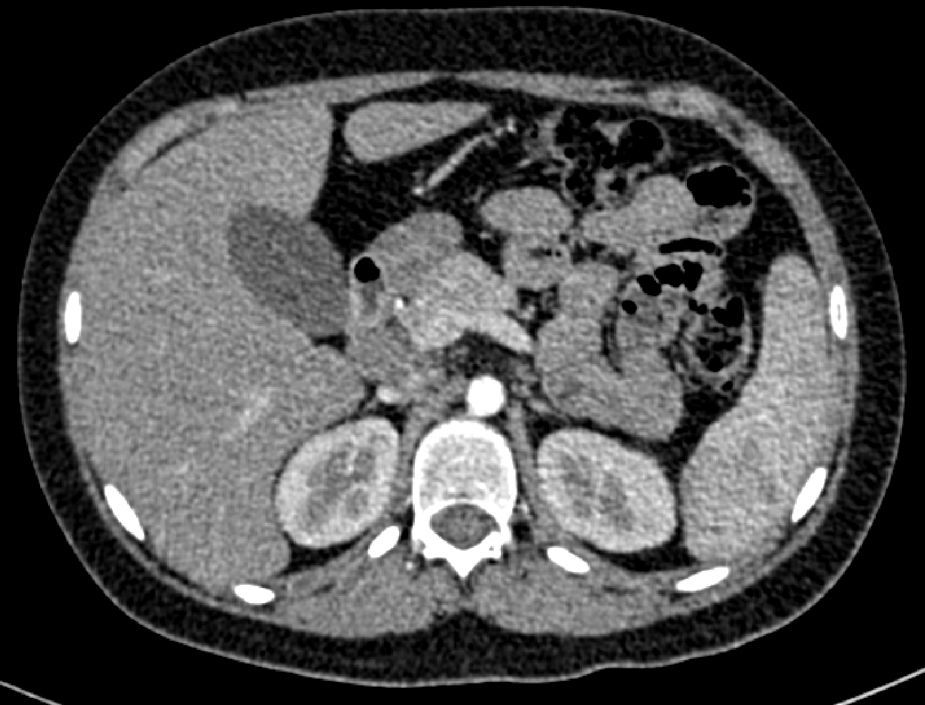
En la tomografía computada de tórax y abdomen se observaron conglomerados ganglionares en mediastino, mesenterio, retroperitoneo y región inguinal, infiltrado micronodular difuso bilateral, hepatomegalia, esplenomegalia, así como litiasis renal derecha (fig. 3).

En el ultrasonido renal se observaron ambos riñones de tamaño y morfología normal, relación córtico-medular adecuada sin evidencia de dilatación de los sistemas colectores. En el riñón derecho se identificó, a nivel del seno, imagen redondeada hiperecogénica de 6.7mm compatible con lito, adecuada perfusión a la aplicación de Doppler. Vejiga moderadamente distendida sin datos de obstrucción.
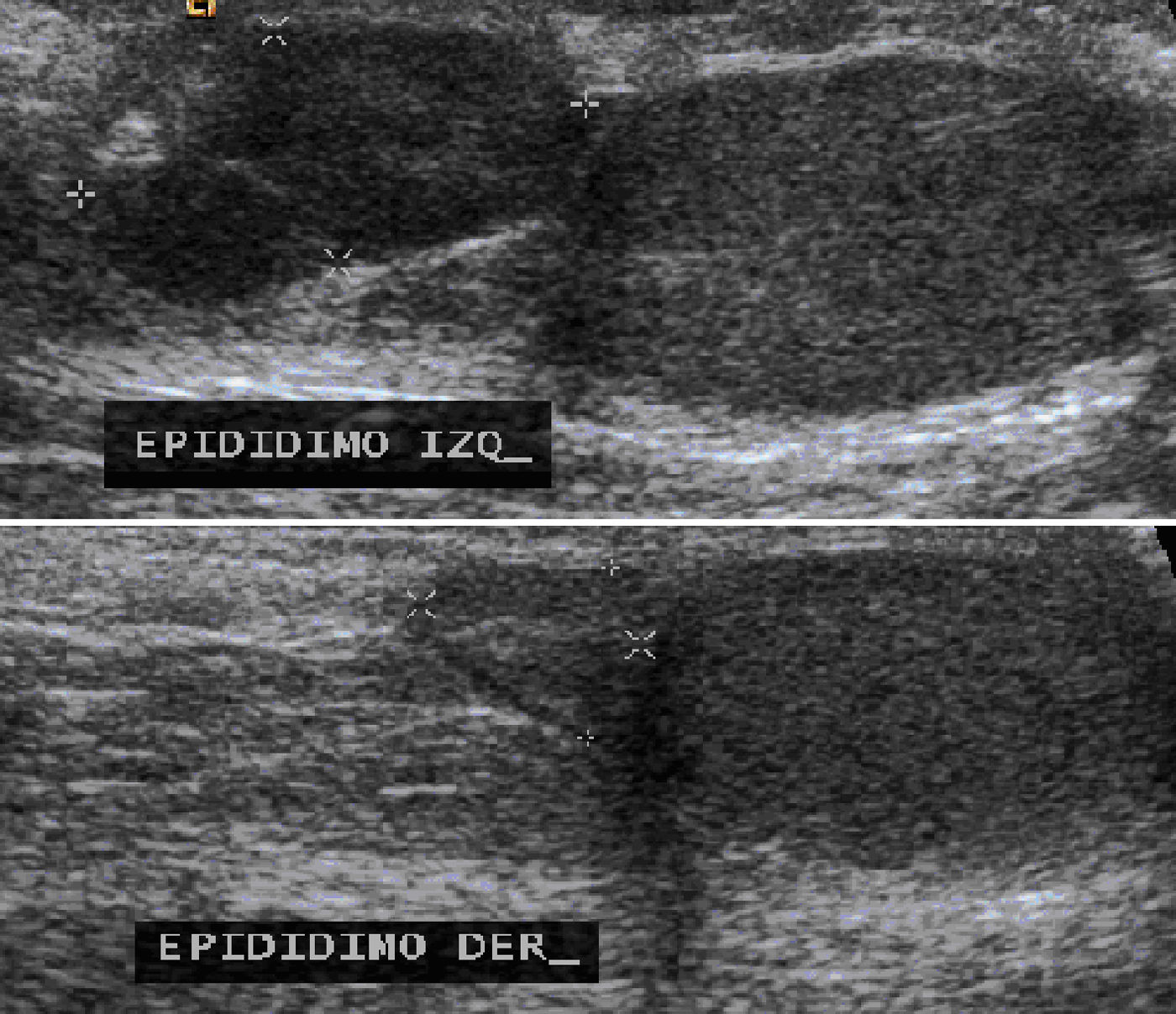
El ultrasonido testicular mostró ambos testículos y epidídimo derecho de tamaño normal, pero el epidídimo izquierdo se observó aumentado de tamaño, de bordes lobulados y bien definidos con dimensiones de 15x7x15mm y con vascularidad central en la modalidad Doppler (fig. 4).

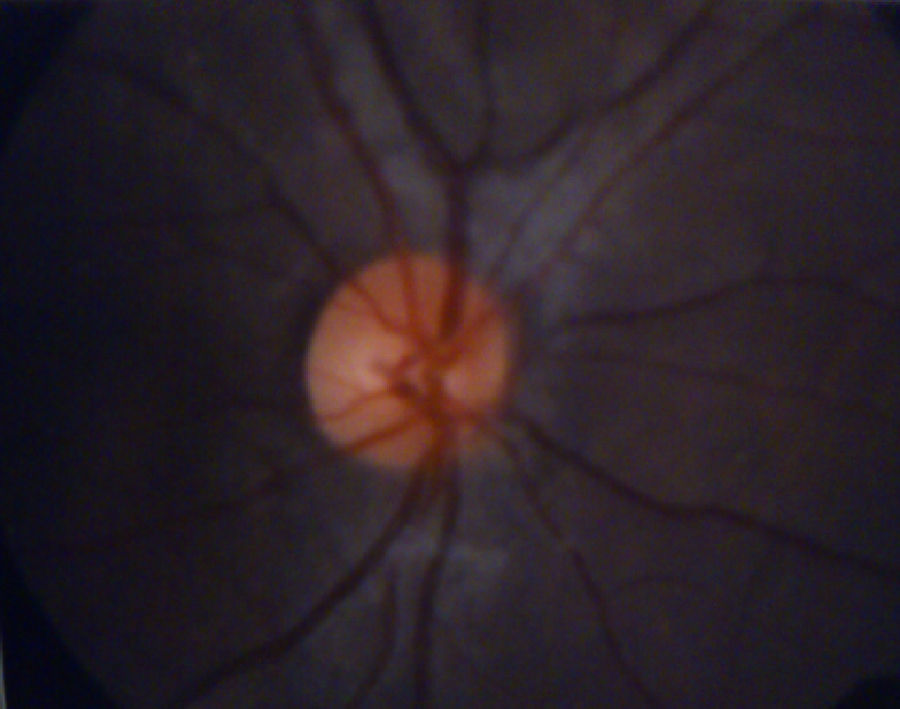
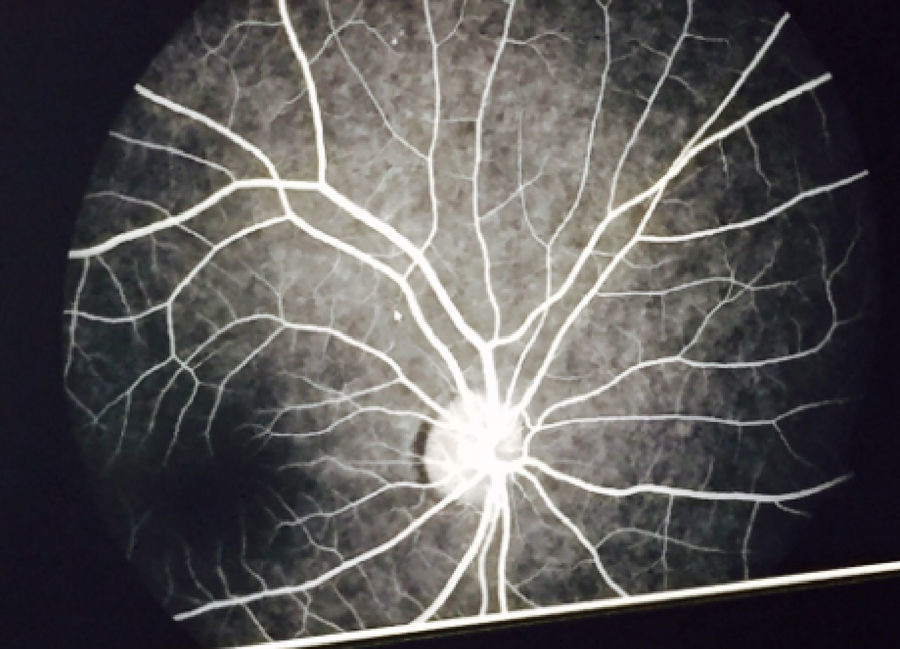
El examen de fondo de ojo mostró periflebitis temporal y nasal inferior, compatible con alteraciones vasculares de sarcoidosis (fig. 5). Sin embargo, la fluorangiografía retiniana reportó, en ambos ojos, nervios ópticos con excavación 3/10, emergencia central de vasos, retinas aplicadas, fase coroidea normal, sin alteraciones en los trayectos vasculares (fig. 6).


En el ecocardiograma se observó situs solitus, levoapex, concordancia AV y VA normal, FEVI 72%, FA 40%, PSVD 17mmHg, septum interventricular en diástole de 9mm.
Las pruebas de función pulmonar (grado de calidad A, cumplió con criterios de aceptabilidad y repetibilidad de la ATS) dieron los siguientes resultados. Espirometría: VEF1/FVC 89 (102), FVC 2.73 (91%), VEF1 2.43 (93%); pletismografía: SVC 2.72 (90), IC 1.85 (74), ERV 0.87 (164), TGV 1.27 (71), VR 0.40 (45), TLC 3.11 (73) RV/TCL 13 (57), compatible con patrón restrictivo leve.
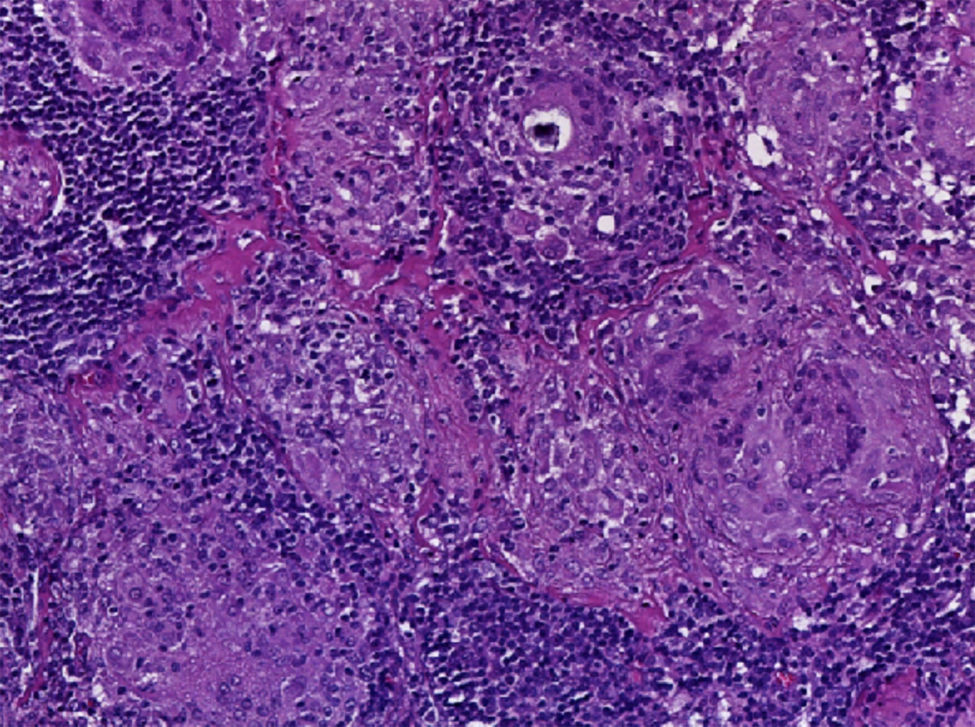
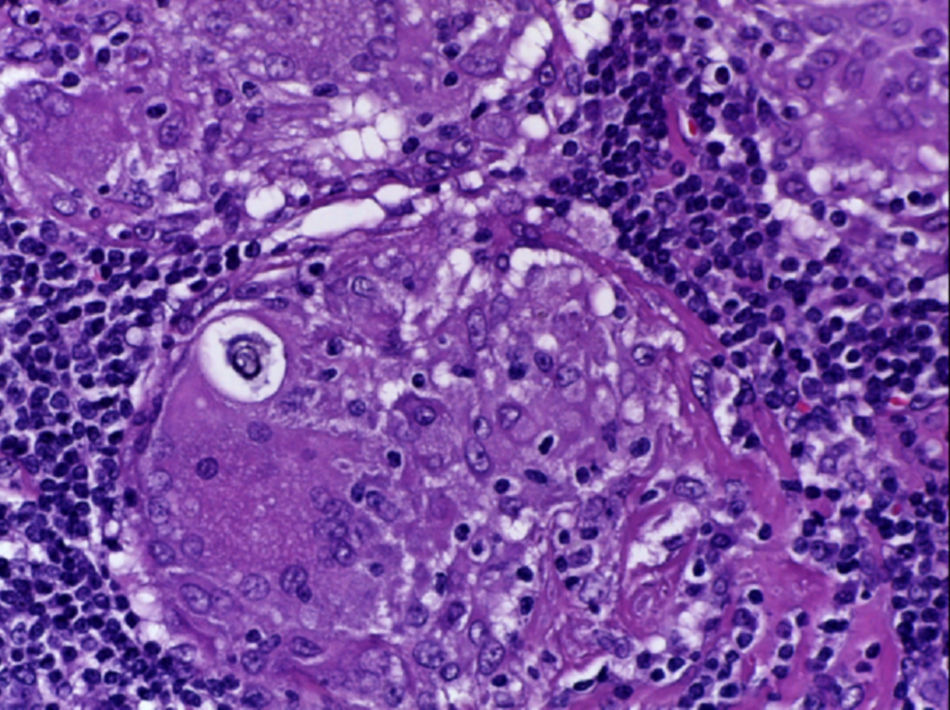
Se realizó la biopsia de ganglio supraclavicular con tinciones de hematoxilina/eosina, Grocott, Ziehl-Neelsen y PAS. Los cortes histológicos mostraron ganglios linfáticos con pérdida de la arquitectura, secundaria a la formación de múltiples granulomas no caseificantes confluentes entre sí (fig. 7), constituidos por numerosos macrófagos epitelioides, células gigantes tipo Langhans y otras tipo cuerpo extraño. Se observaron, de forma ocasional, calcificaciones concéntricas o cuerpos de Schaumann (fig. 8) y formas compatibles con cuerpos asteroideos (fig. 9). Con las tinciones especiales no se observaron microorganismos, y las pruebas de PCR así como los cultivos en tejido del ganglio para Mycobacterium tuberculosis fueron negativos. Diagnóstico histopatológico: ganglios linfáticos supraclaviculares con linfadenitis crónica granulomatosa no caseificante concordante con sarcoidosis.



En los primeros días de estancia hospitalaria, el paciente presentó cefalea y signos de Kernig y Brudzinski, por lo cual se realizó tomografía computada de cráneo que mostró adecuada diferenciación de sustancias gris y blanca, sistema ventricular y estructuras vasculares normales, sin alteraciones meníngeas. Líquido cefalorraquídeo (LCR) trasparente, coagulación y película negativas, proteínas 30 mg/dl, glucosa 49mg/dl, leucocitos 1/campo. Tinción de Gram y cultivos negativos. Prueba de PCR para Mycobacterium tuberculosis en LCR negativa.
Se inició tratamiento con prednisona 2mg/kg/día e hidroclorotiazida 1.2mg/kg/día, con lo cual se observó mejoría clínica de su estado general, con remisión de la hepatomegalia y la esplenomegalia, del eritema nodoso, así como de las adenopatías y de la masa testicular. Posteriormente, desaparecieron las adenopatías mediastinales y los infiltrados pulmonares (fig. 10).

Cuatro meses después el paciente estaba asintomático, había recuperado 15kg de peso y se había incorporado a sus actividades escolares. La tomografía de abdomen sin adenopatías retroperitoneales. De igual forma, el ultrasonido renal no mostró lesiones focales ni dilatación de sistemas colectores y sin evidencia de litiasis renal. Se repitieron las pruebas de espirometría y pletismografía que fueron compatibles con patrón normal. Los exámenes de laboratorio mostraron calcio sérico 9.0mg/dl, fósforo 5.8mg/dl, creatinina 0.8mg/dl, ácido úrico 6.2mg/dl y calciuria en orina de 24 horas 0.93mg/kg/día. Así mismo, la determinación de control de la ECA fue de 71 U/l, por lo que se consideró que la sarcoidosis se encontraba en remisión.
3DiscusiónEl presente caso corresponde a un adolescente de sexo masculino con un cuadro clínico compatible con sarcoidosis infantil de inicio tardío, con grave afección del estado general que le llevó a la caquexia. También presentaba daño pulmonar, hepatomegalia, esplenomegalia, adenopatías múltiples, eritema nodoso y masa escrotal. Se realizó el protocolo de abordaje diagnóstico para pacientes con hepato-esplenomegalia, el cual permitió descartar varias enfermedades infecciosas, como tuberculosis, micosis sistémicas, sífilis e infección por virus Epstein Barr (VEB) y virus de la inmunodeficiencia humana (VIH), además de otras causas de síndrome infiltrativo, como leucemias, linfomas, tumores metastásicos y linfohistiocitosis hemofagocítica, mediante los estudios de tomografía computada y las biopsias de médula ósea y de ganglio linfático. Se descartaron, incluso, vasculitis sistémicas y otras enfermedades autoinmunes, así como los tumores testiculares de la adolescencia con base en los hallazgos del ultrasonido testicular y con los niveles normales de marcadores tumorales como alfa-feto proteína y beta-gonadotropina61.
El diagnóstico de sarcoidosis se sospechó ante la presencia de un padecimiento sistémico con hipercalcemia, hipercalciuria y litiasis urinaria, acompañadas de elevación de los niveles de la ECA y del sIL-2R. Se comprobó, mediante el estudio histopatológico de la biopsia de ganglio linfático, la presencia de múltiples granulomas no caseificantes conformados por macrófagos epitelioides, células gigantes tipo Langhans, y la presencia de cuerpos de Schaumann y cuerpos asteroideos. Sin embargo, a su ingreso, no se pudo realizar el análisis citológico del lavado broncoalveolar ni se pudo aplicar la prueba de Kveim-Siltzbach.
A su ingreso se detectó insuficiencia respiratoria crónica tipo I, sin repercusión cardiovascular, con hipoxemia, normocapnia e hiperlactatemia. La radiografía de tórax mostró infiltrado micronodular y reticular difuso con adenopatías hiliares bilaterales, concordante con el Estadio II de la clasificación de la American Thoracic Society para sarcoidosis torácica. Así mismo, la espirometría y la pletismografía reportaron una disminución de la relación VEF1/FVC, compatible con una neumopatía de patrón restrictivo leve como se ha descrito en las dos primeras etapas de la sarcoidosis pulmonar, que se normalizó después del tratamiento con prednisona.
Durante su estancia hospitalaria, el paciente también presentó cefalea y signos de irritación meníngea que se atribuyeron a la infiltración sarcoidea de las meninges, ya que el análisis citoquímico y los estudios microbiológicos en el LCR así como la tomografía computada de cráneo se reportaron normales. Estas alteraciones clínicas también desaparecieron con el tratamiento62,63.
A pesar de que el paciente nunca presentó síntomas visuales, se realizó el examen oftalmológico debido a la alta frecuencia de afecciones oculares en sarcoidosis. Se encontraron hallazgos como periflebitis compatible con la afección del tracto uveal por sarcoidosis, que no se documentaron en el estudio de fluorangiografía retiniana posiblemente porque esta se realizó varios días después de haber iniciado el tratamiento esteroideo. Como ya ha sido descrito previamente, este es el tratamiento de elección ya que, en la mayoría de los casos, mejora rápidamente las lesiones oculares de la sarcoidosis, evitando la progresión de la enfermedad y la aparición de otras lesiones irreversibles que conduzcan a la ceguera64.
La hipercalcemia en sarcoidosis tradicionalmente se ha atribuido a la producción de calcitriol por los macrófagos y monocitos activados de los granulomas, lo que a su vez incrementa la absorción intestinal del calcio y la actividad de los osteoclastos, con resorción ósea y aumento en los niveles de calcio sérico. Sin embargo, en el presente caso, la determinación de calcitriol (1,25 dihidroxi-vitamina D3) se encontró dentro del rango normal, y la PTH por debajo de los niveles normales; esto indica, por un lado, que existía inhibición de las glándulas paratiroideas debido al nivel elevado de calcio sérico y, por otro, que probablemente la hipercalcemia se debió a otro factor autónomo, como la producción del PTHrP por los macrófagos activados, lo que no fue posible medir en este caso65.
Inicialmente presentaba insuficiencia renal aguda, que remitió sin terapia de reemplazo renal ya que no tuvo criterios para diálisis. Además, se comprobó hipercalciuria hipercalcémica acompañada de hiperfosfaturia y disminución de la reabsorción tubular de fosfatos, que se expresó en forma de oxalato de calcio y fosfato de calcio, lo cual le ocasionó enfermedad litiásica renal. Si bien la presentación clínica de dolor cólico agudo es poco habitual en la urolitiasis de los niños, este paciente se presentó con dolor en flanco derecho acompañado de hematuria microscópica. Como parte del manejo médico se proporcionó tratamiento con hidroclorotiazida, con el fin de reducir la excreción renal de calcio y disminuir la formación de litiasis cálcica de oxalato y fosfato de calcio, logrando la remisión de la hipercalciuria, la expulsión del cálculo renal, y por lo tanto la disminución del riesgo litogénico66–72 (fig. 11).

En este paciente no se realizaron estudios de antígenos de histocompatibilidad (HLA). En cuanto a la función inmunológica, se observó hipergammaglobulinemia policlonal moderada, con niveles adecuados de C3 y C4, hallazgo ya descrito en pacientes con sarcoidosis. Así mismo, las pruebas de función fagocítica con reducción de nitroazul de tetrazolio y quimioluminiscencia resultaron normales, por lo cual se descartó la posibilidad de enfermedad granulomatosa crónica.
Si bien la citometría de flujo en sangre periférica reportó un número total de linfocitos dentro del rango normal, la identificación de las principales subpoblaciones linfocitarias mostró el 50.7% de linfocitos T, el 33.4% de linfocitos B y el 13.7% de células NK, con una relación CD4/CD8=1.22. Lo anterior podría explicar la paradoja inmunológica observada en las pruebas cutáneas, ya que se consideró que el paciente presentaba inmunidad celular y humoral normal73–77.
Finalmente, se debe señalar que la sarcoidosis infantil es un padecimiento poco diagnosticado en México, por lo cual no existe información clínica ni epidemiológica sobre la enfermedad en la población. Se considera que el presente caso puede contribuir al conocimiento de la sarcoidosis en los niños, en quienes los síntomas clínicos son inespecíficos. En los pocos casos reportados, ha sido confundida con otras enfermedades como la artritis idiopática juvenil, la tuberculosis o con micosis sistémicas. Sin embargo, el compromiso de múltiples órganos con riesgo de presentar complicaciones que pueden conducir a discapacidades graves y/o a la muerte, obliga a considerarla en el abordaje diagnóstico de los pacientes con adenopatías generalizadas, hepatomegalia y esplenomegalia. Esto con la finalidad de iniciar precozmente la terapia con glucocorticoides, que es el tratamiento más eficaz para evitar la progresión de la enfermedad en la mayoría de estos pacientes, y que es fácilmente asequible en nuestro medio.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.