


Paciente de sexo femenino referida en 2008, a los 5 meses de edad, al Hospital Infantil de México Federico Gómez (HIMFG) con diagnóstico de catarata congénita bilateral.
1.1Antecedentes heredofamiliaresMadre de 29 años, dedicada al hogar, y padre de 35 años, campesino. Medio hermano de 12 años. Probable consanguinidad entre padre y madre.
1.2Antecedentes no patológicosTodos aparentemente sanos y residentes del Estado de México.
1.3Antecedentes perinatales y patológicosLa madre tenía antecedente de un aborto espontáneo de etiología desconocida. Fue producto del tercer embarazo; recibió hierro, ácido fólico y vacunación con toxoide tetánico, se realizó cuatro ultrasonidos, el último de los cuales reportó microcefalia. Nació de término por cesárea iterativa. Pesó 3,180g, calificación de Apgar 9/9 y se dio de alta con la madre.
Se encontró con microcefalia, hipotonía central y retraso en el desarrollo psicomotor. Los potenciales auditivos indicaron hipoacusia moderada bilateral. En los estudios de imagen se observó hiperdensidad de la sustancia blanca en el centro semioval y periventricular bilateral, con calcificaciones aisladas y un quiste aracnoideo retrocerebeloso.
Enero de 2009. Se realizó papuloplastía y vitrectomía. Ese mismo año se hizo funduplicatura y gastrostomía por alteración en la mecánica de la deglución y se indicó plan de alimentación con licuados de 900kcal.
Septiembre de 2010. Se solicitó perfil genético y TORCH. El cariotipo fue 45XX. Se realizó el diagnóstico de síndrome de Cockayne.
Mayo de 2011. Fue hospitalizada por neumonía. Durante este internamiento recibió dos transfusiones de concentrado eritrocitario por anemia.
Mayo de 2013. Acudió al servicio de Urgencias por edema de manos y pies, irritabilidad, distensión abdominal y ausencia de evacuaciones. Se diagnosticó neumonía adquirida en la comunidad. Se encontró con hemoglobina de 4.8mg/dl y hematocrito de 16% con datos de choque, por lo que se transfundió concentrado eritrocitario. Se inició manejo con aminas durante 24h y cobertura antimicrobiana con cefepime y amikacina. Se egresó por mejoría.
1.4Padecimiento actualAcudió al Servicio de Urgencias por presentar datos de dificultad respiratoria y deterioro neurológico. Al ingreso se encontró con inestabilidad hemodinámica, con datos de bajo gasto, llenado capilar en 4 s, pulsos débiles y taquicardia. Se administraron cuatro cargas de solución fisiológica a 20ml/kg y se intubó con cánula número 4.5. Persistió con respuesta inflamatoria sistémica y acidosis metabólica, por lo que se administraron dos cargas más de solución fisiológica e infusión de adrenalina a 0.3μg/kg/min.
Gasometría arterial. pH 7.22, pO2 64mmHg, pCO2 40.5mmHg, saturación de oxígeno 87.6%, HCO3 16.2 mEq/l, lactato 1.8mmol/l, diferencia arteriovenosa de oxígeno 5.58, índice de Kirby 64.
Biometría hemática. Leucocitos 15,700/μl, neutrófilos 67%, bandas 10%, linfocitos 1%, hemoglobina 8.2g/dl, hematocrito 25%, plaquetas 388,000/μl.
Química sanguínea. Nitrógeno ureico 34mg/dl, creatinina 1.3mg/dl, glucosa 89mg/dl, deshidrogenasa láctica 524 U/l, fósforo 5.2mg/dl, calcio 6.3mg/dl, sodio 139 mEq/l, potasio 4.6 mEq/l, cloro 105 mEq/l, bilirrubina total 0.92mg/dl, bilirrubina directa 0.55mg/dl.
Tras la aparición de taquiarritmia se cambió la adrenalina por dobutamina y se inició con infusión de amiodarona a 5μg/kg/min. Cursó con hipoxia persistente, reserva venosa de 17%, mala perfusión y evento de desaturación de hasta el 39% a pesar del apoyo ventilatorio. Finalmente presentó bradicardia y asistolia. Se iniciaron maniobras avanzadas de reanimación con 10 ciclos de compresiones, cuatro dosis de adrenalina y una dosis de bicarbonato. Presentó salida de sangre fresca por cánula endotraqueal, y se declaró la muerte.
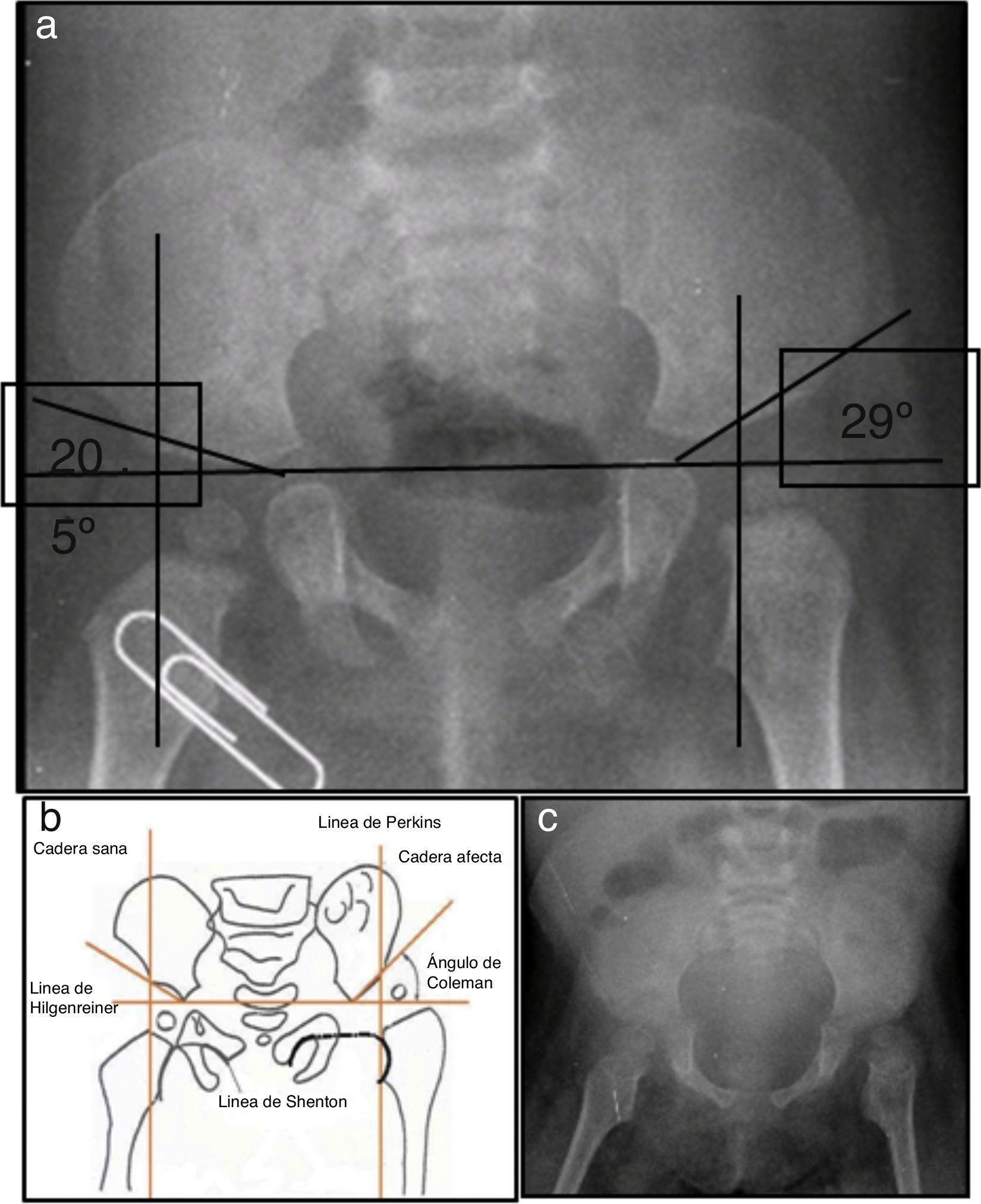
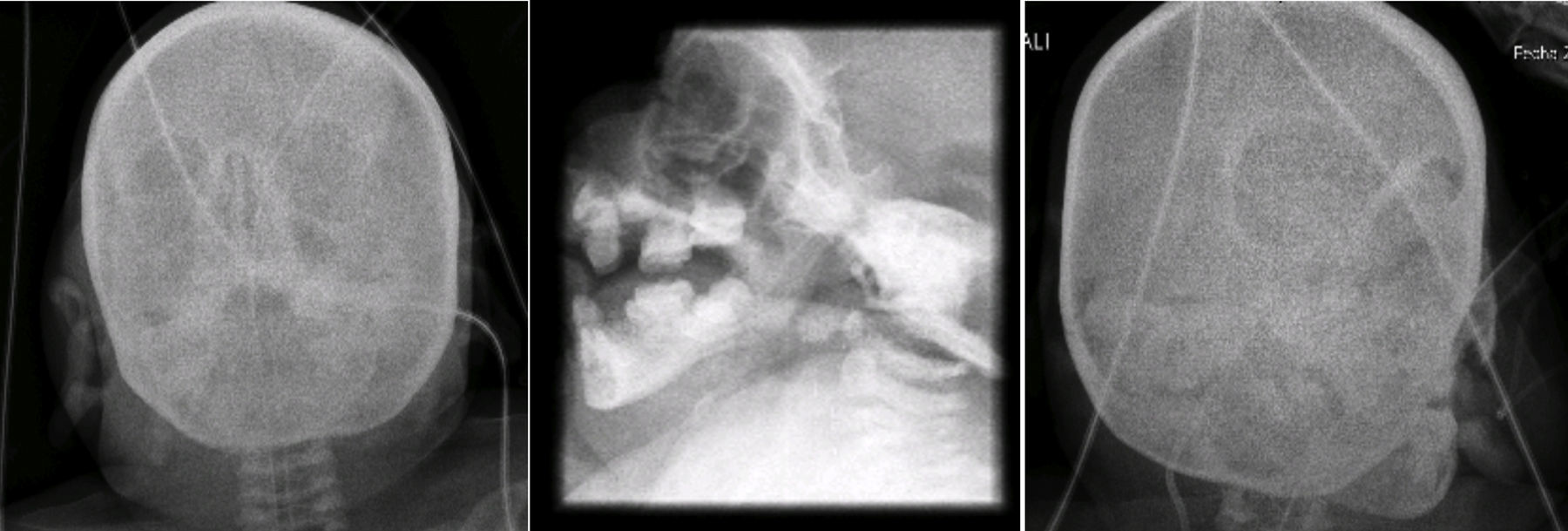
2Presentación del caso2.1Imagenología (Dra. Pilar Diez)En la radiografía de pelvis de 2009, al año de edad, se observó displasia de cadera izquierda (Figura 1A). Los ángulos acetabulares deben medir de 28° a 29° al nacimiento (Figura 1B). Al año de edad, este ángulo debería haber disminuido a menos de 22°, como se observa en cadera derecha, aunque no así en la izquierda que tiene 29°. Además, la cabeza femoral debería estar en el cuadrante interno e inferior, lo que no sucede en el caso de la cadera izquierda. La morfología de la pelvis está alterada, es plana, no se observan los agujeros obturadores y hay agenesia de coxis (Figura 1C).

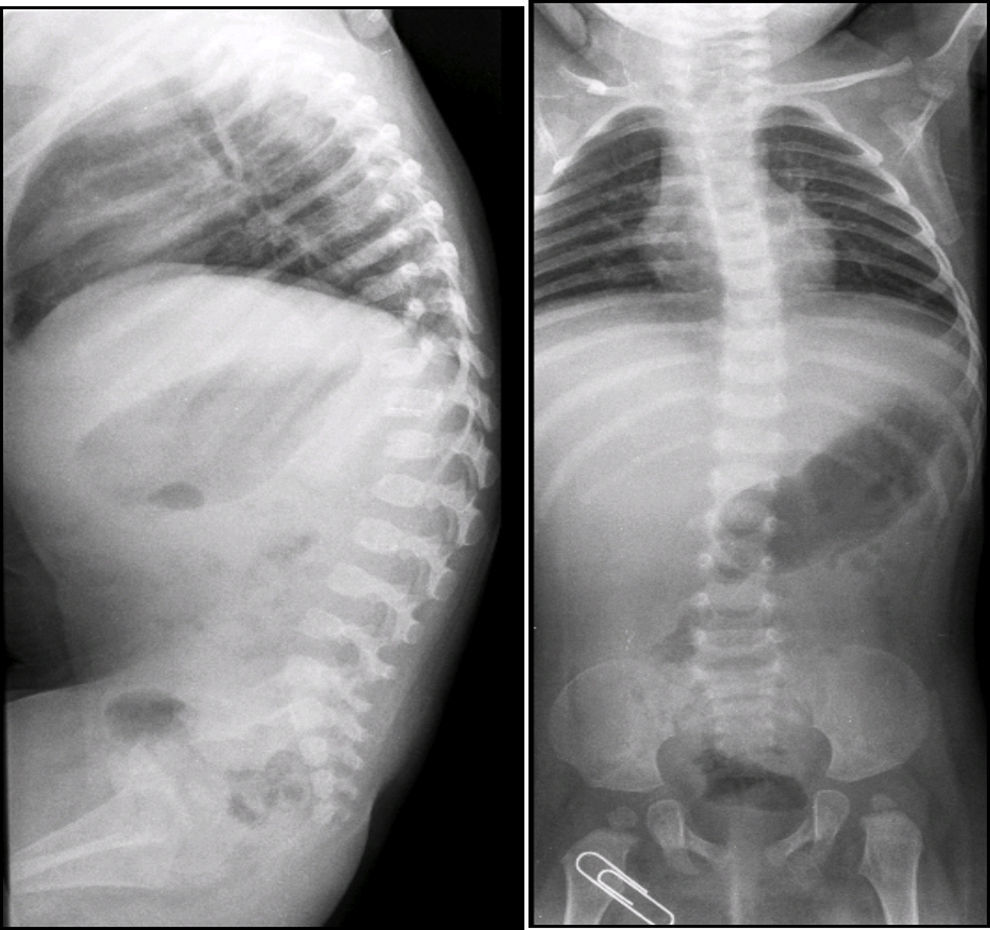
En las radiografías de columna lateral se observó escoliosis toráxica levoconvexa y xifosis toracolumbar con alteraciones en la morfología vertebral (Figura 2). La curvatura toráxica es más acentuada de lo normal para la edad de la paciente. La xifosis toracolumbar se explica por la falta de deambulación de la paciente. Los cuerpos vertebrales están discretamente aplanados, alargados, con muescas y los espacios intersomáticos están muy aumentados. También se observan alteraciones del sacro y del coxis.

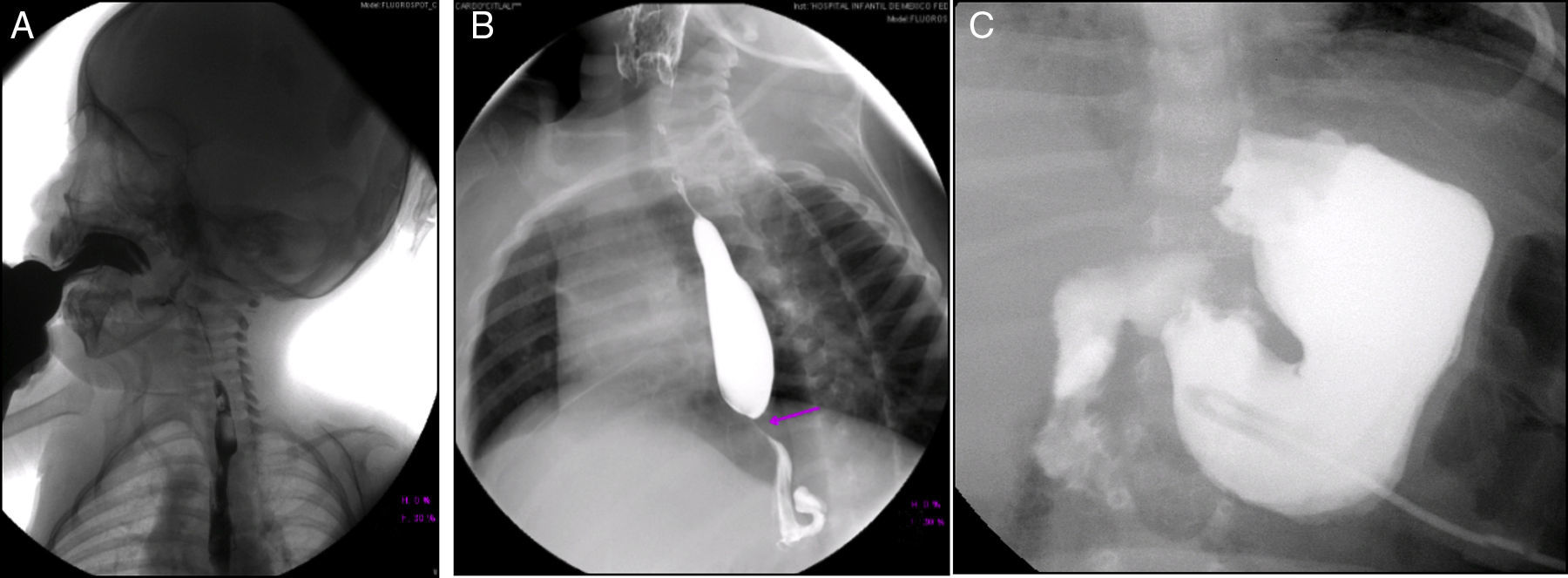
En la serie esófago-gastroduodenal (Figura 3) se observó disfunción velo palatina con reflujo nasofaríngeo (Figura 3A). En las siguientes imágenes se observan cambios postquirúrgicos por una funduplicatura discretamente apretada que condicionó la disminución en el calibre del esófago en su tercio distal (Figura 3B), y en la última se observa la sonda de gastrostomía (Figura 3C).
 Reflujo nasofaríngeo (2009); B) cambios por funduplicatura (2011); C) sonda de gastrostomía (2012).")
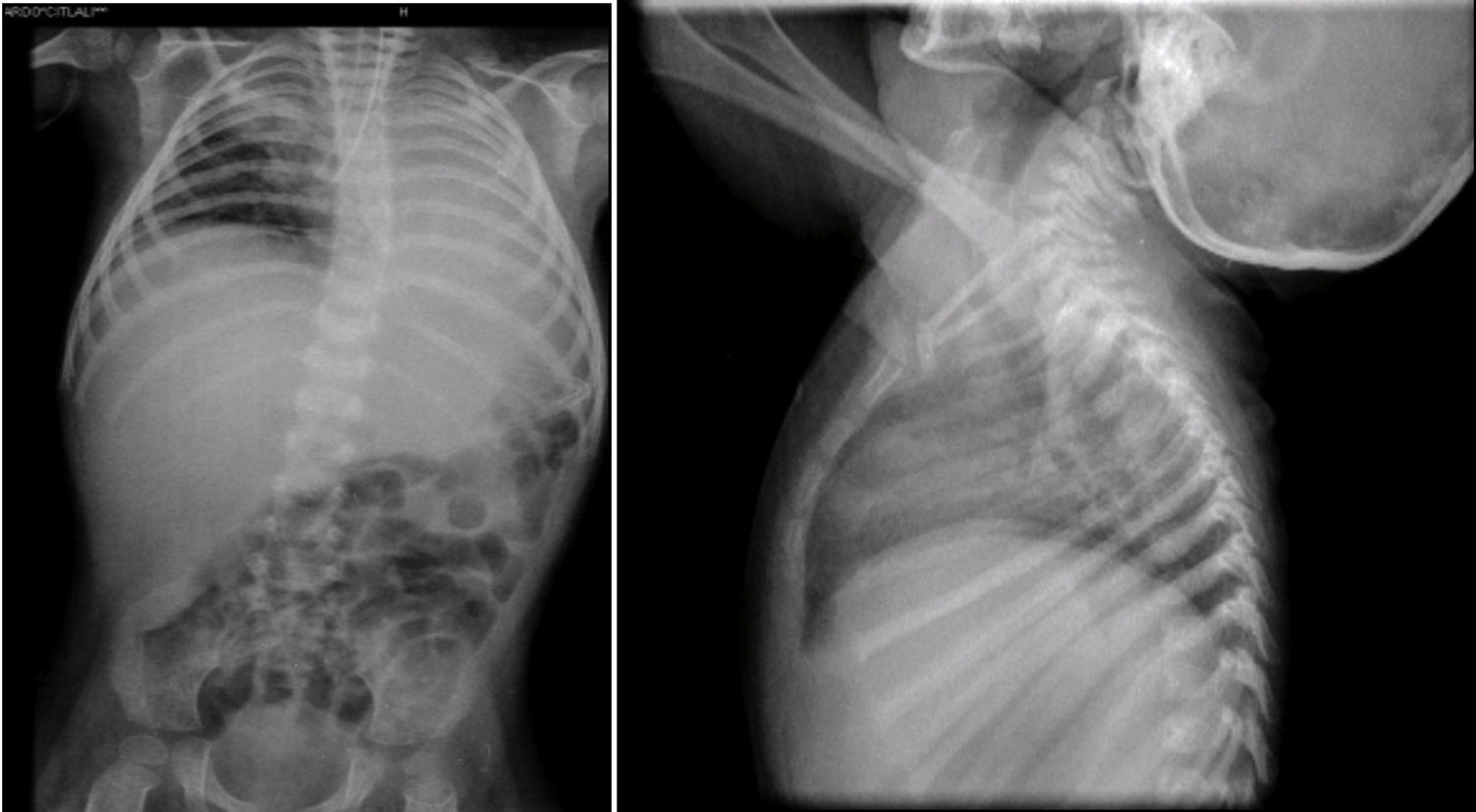
En la radiografía toracoabdominal se observó un infiltrado retículo nodular del lado derecho y atelectasia izquierda, probablemente secundaria a localización de la cánula endotraqueal en el bronquio derecho (Figura 4). También se observó hepatomegalia y los cuerpos vertebrales alterados. Las costillas y clavículas se ven muy adelgazadas, con esclerosis en los bordes. Estas imágenes son características del síndrome de Cockayne.

En las radiografías de cara y cráneo se observó puente nasal con forma de gancho, e hipertelorismo. Al paso de los años, la cara de la paciente pierde tejido adiposo. La silla turca no se observa alterada (Figura 5).

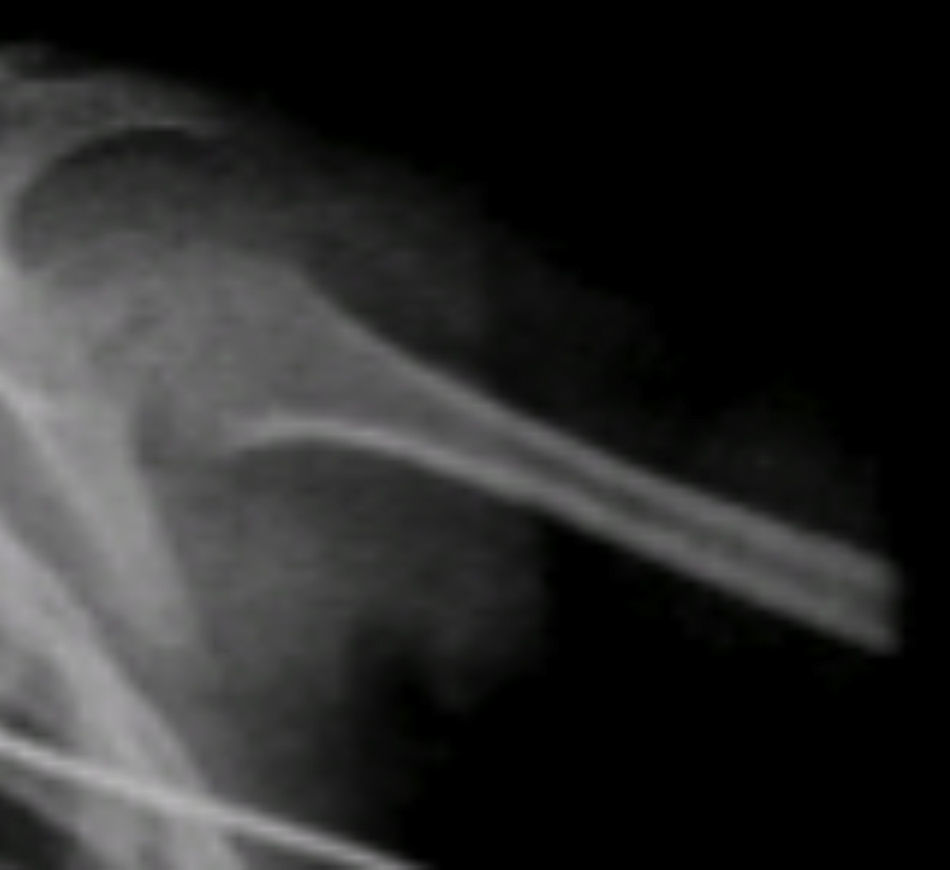
Se observaron otros hallazgos como las metáfisis cortas y anchas y las diáfisis muy delgadas y osteopénicas, con zonas de esclerosis (Figura 6).
3Patología (Dr. Carlos Alberto Serrano)
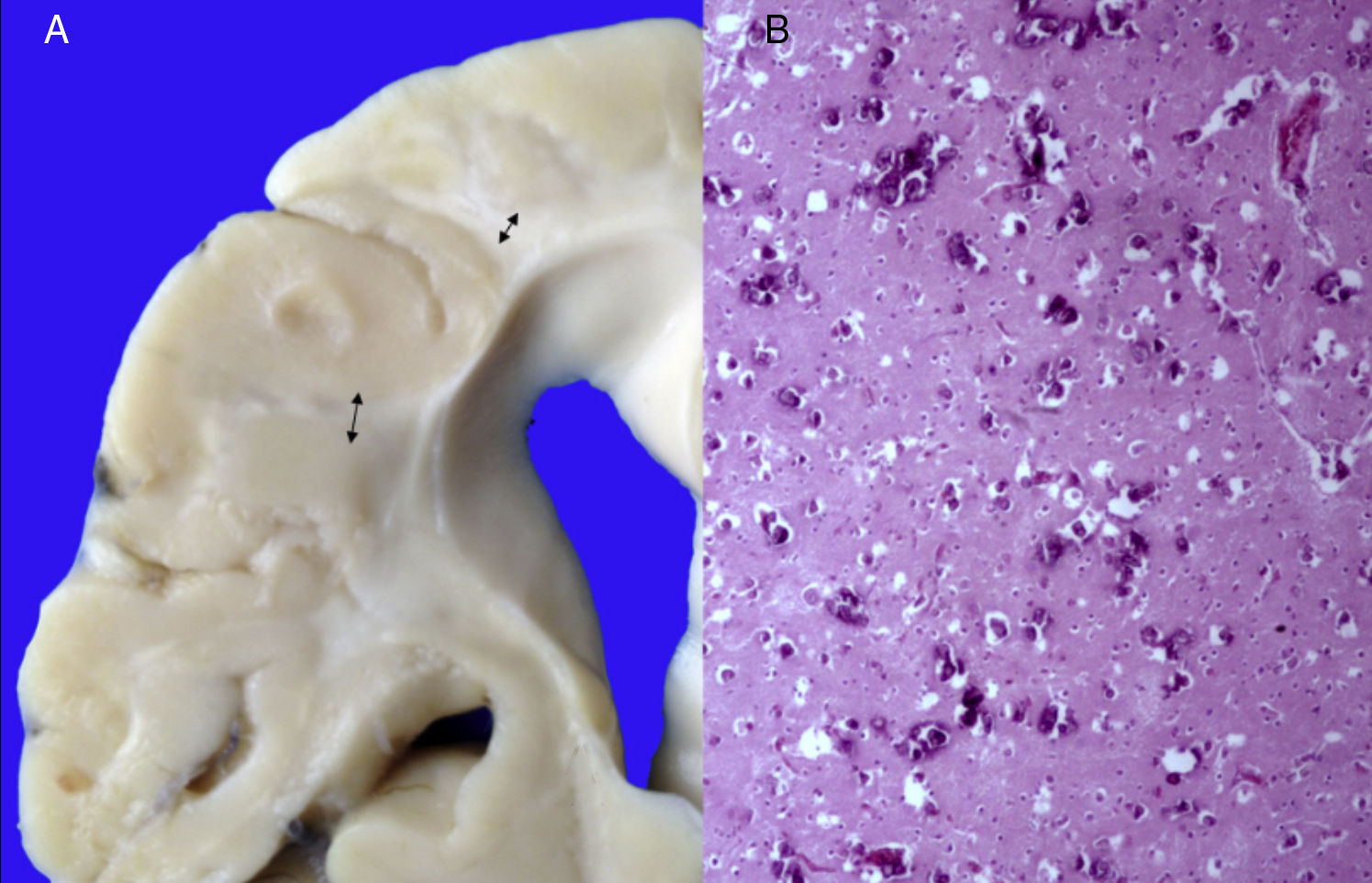
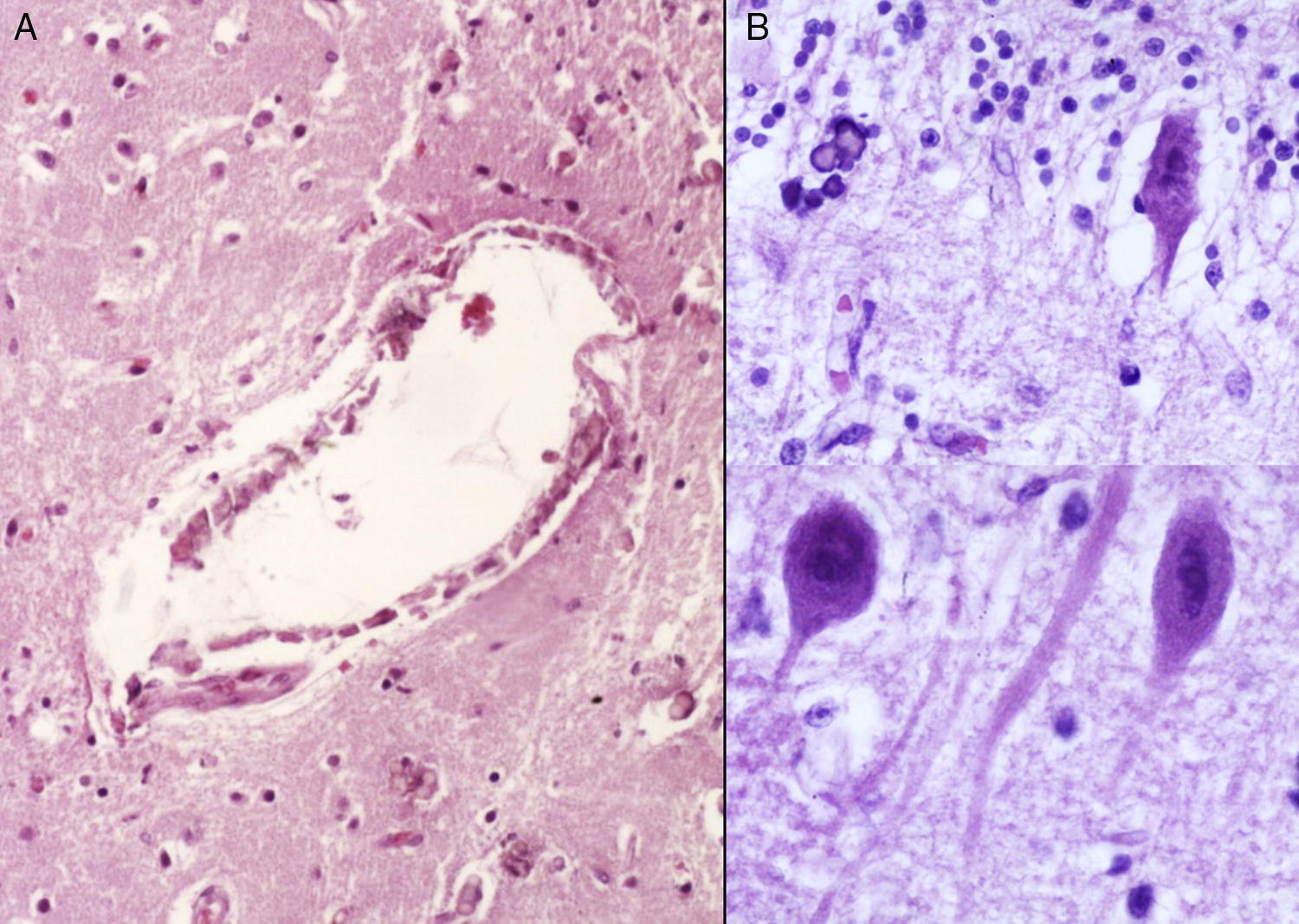
Los hallazgos fenotípicos que presentaba el cuerpo fueron facies progeroide, cabello delgado y reseco, encéfalo con múltiples calcificaciones cerebrales, microcefalia (con un peso del encéfalo de 300g contra un esperado de 1,200g) y atrofia cerebral. Los cortes sagitales del cerebro mostraron características típicas del síndrome de Cockayne: pérdida casi total de la sustancia blanca (únicamente se observaba la sustancia gris, lo que le daba un aspecto delgado y alargado a las circunvoluciones), con surcos amplios y discreta dilatación del sistema ventricular. Se observaron áreas de color pardo que correspondían a microcalcificaciones. En los cortes histológicos de los núcleos basales se observaron múltiples calcificaciones en el parénquima cerebral y pérdida de cuerpos neuronales. Las neuronas residuales presentaban núcleos picnóticos con disminución del citoplasma, signos de muerte celular anticipada, al igual que las células de Purkinje del cerebelo (Figura 7). En un acercamiento se observaron áreas mal delimitadas, de color blanco y aspecto granular, que corresponden a calcificaciones en diversas regiones de la corteza cerebral. En los cortes histológicos se observaron áreas blancas que corresponden a la pérdida de mielina, lo cual se corroboró con la tinción luxol-fast-blue. Existen dos teorías para la pérdida de la sustancia blanca en este síndrome. La primera, por la degeneración de las células que producen la mielina; la segunda, porque ocurren alteraciones vasculares en el encéfalo que llevan a la muerte de estas células. Las características histológicas que se observan en estos pacientes son similares a aquellas que se observan en pacientes de mayor edad que tienen engrosamiento de las paredes arteriales, e incluso ateromas. En la paciente no se observaron estos cambios, pero sí alteraciones en la morfología de las células endoteliales que propiciaban una disminución en la irrigación del parénquima cerebral (Figura 8).
 Encéfalo con pérdida importante de la sustancia blanca (flechas). B) Corteza cerebral con múltiples calcificaciones.")
 Vasos de la corteza cerebral con calcificaciones de su pared. B) Degeneración roja de las neuronas con microcalcificaciones.")
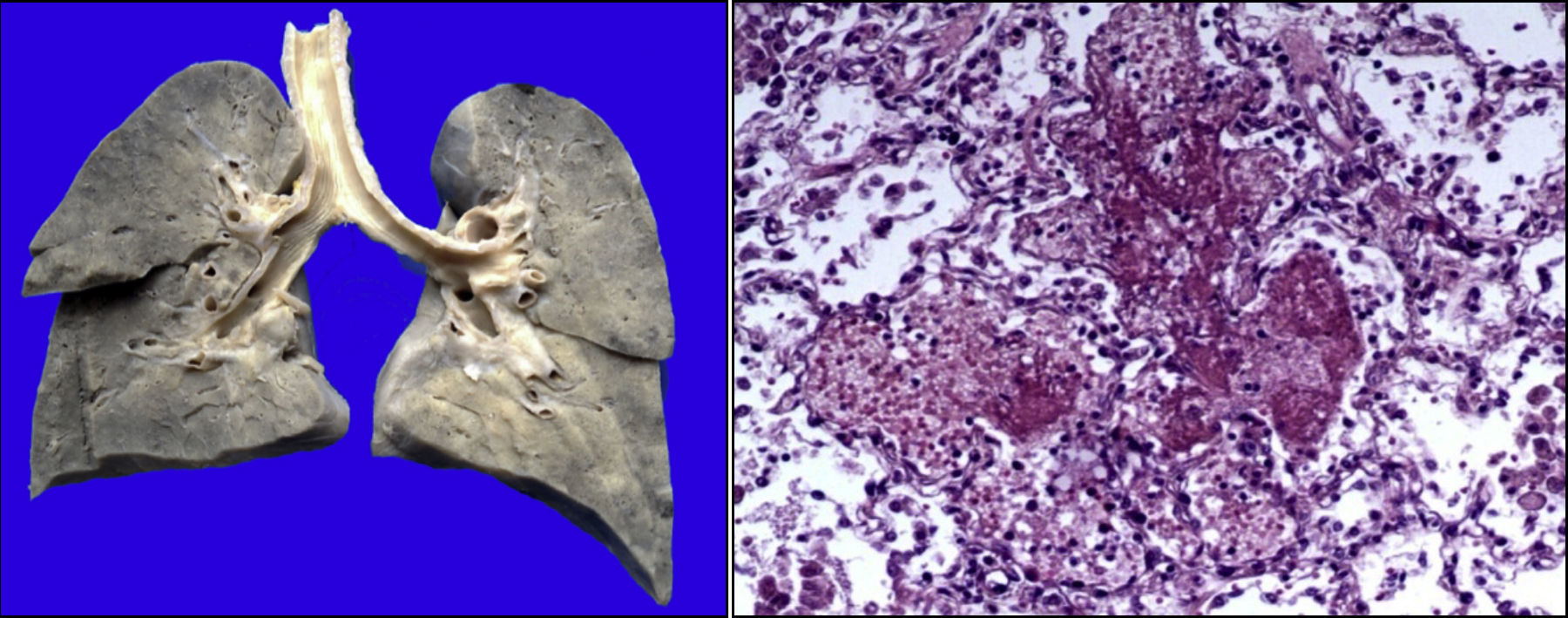
Existía derrame pleural en el lado izquierdo de aproximadamente 75ml. Más del 80% del parénquima pulmonar presentaba una coloración café oscuro y la tráquea presentaba áreas de congestión en la mucosa. Como se puede observar en los cortes histológicos, estas áreas de color café oscuro corresponden a una importante hemorragia intraalveolar. En otras áreas se observó líquido eosinófilo dentro de los alveolos, dato de edema pulmonar agudo. También había necrosis de los tabiques interalveolares, con formación de membranas hialinas y células gigantes multinucleadas, lo que significa que existía un proceso de broncoaspiración crónico. Así mismo, se observaron escasos linfocitos y macrófagos, lo que corresponde a una neumonía en fase de resolución, aunque con daño pulmonar agudo (Figura 9).
.")
El corazón no presentaba alteraciones morfológicas, pero sí tenía cambios por hipoxia crónica caracterizados por atrofia celular, con citoplasma hipercontraído, vacuolas, pérdida del núcleo y edema intercelular importante.
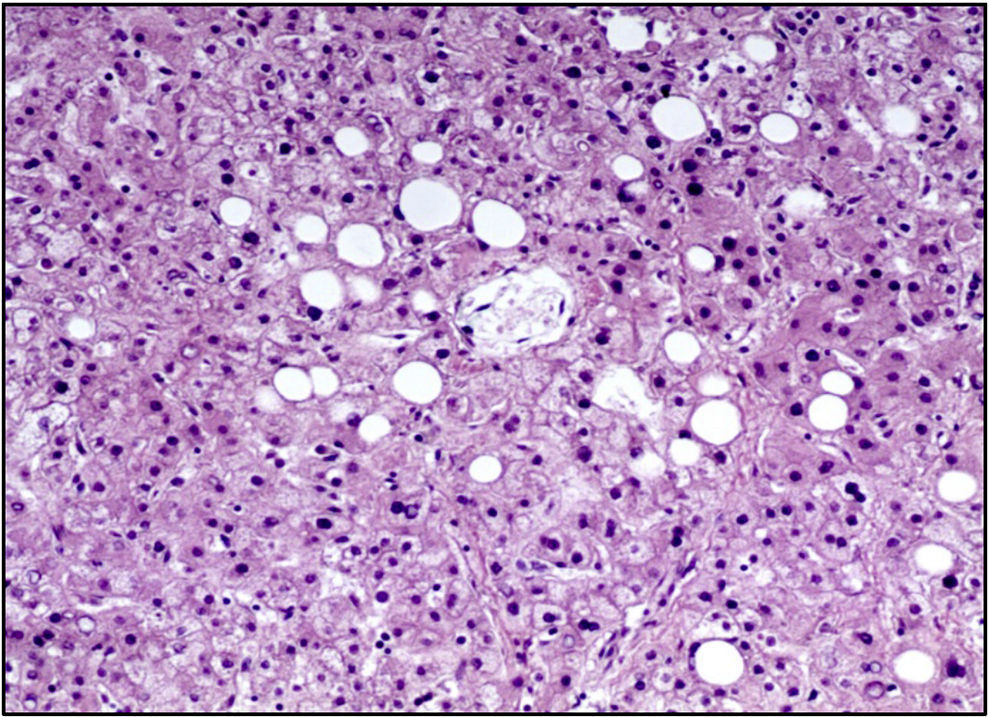
La unión esofagogástrica presentaba engrosamiento de los pliegues y edema en la mucosa. En la microscopía se observaban datos de inflamación crónica, hiperplasia del epitelio y cambios hipóxicos en las capas musculares. El intestino delgado y el colon presentaban únicamente cambios por hipoxia. El hígado presentaba parénquima homogéneo, amarillo y micronodular en algunas áreas. A nivel histológico se apreciaba esteatosis macrovesicular alrededor de las venas centrales, la cual se explica por el estado nutricional y por el estado de choque que presentó al final (Figura 10).

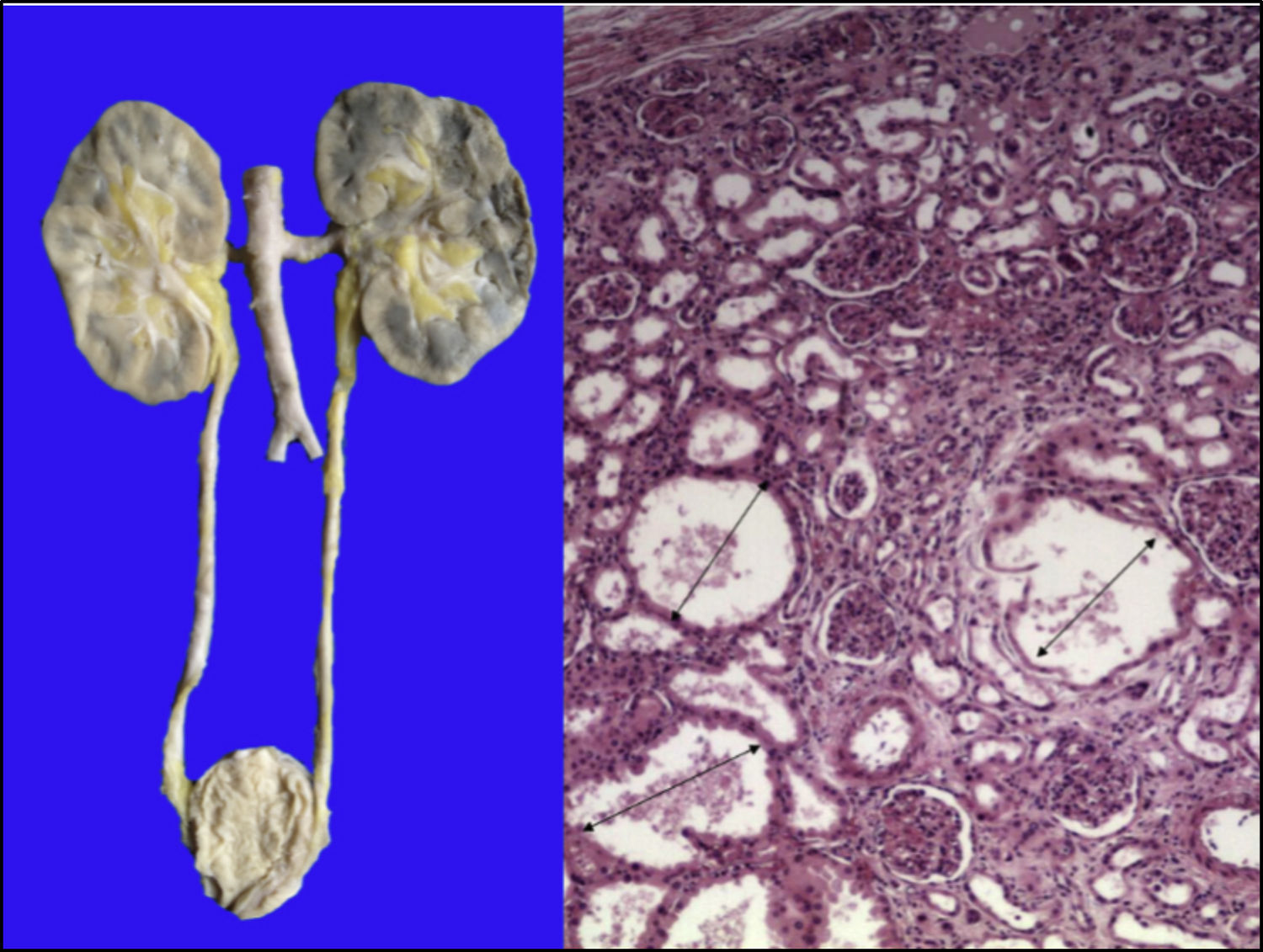
Los riñones se observaron discretamente lobulados por congestión en la médula renal. En la microscopía se ven dilataciones quísticas de los túbulos con algo de material proteínico en su interior, principalmente en la unión corticomedular. Los glomérulos estaban bien conservados (Figura 11).
.")
La medula ósea tenía una celularidad normal. Las tres series hematopoyéticas con maduración adecuada. Los ganglios linfáticos presentaban poco tejido linfoide. En algunos ganglios linfáticos no había formación de folículos secundarios.
3.1Diagnósticos anatómicos finalesEl diagnóstico principal en este caso fue síndrome de Cockayne.
3.2Alteraciones concomitantesLos diagnósticos anatomopatológicos concomitantes fueron los siguientes:
- •
Estado post-funduplicatura y gastrostomía
- •
Neumonía por aspiración en fase de resolución
- •
Hemorragia pulmonar bilateral y daño alveolar difuso extenso
- •
Datos anatómicos de choque: miopatía visceral hipóxico-isquémica en prácticamente todo el tubo digestivo y en el miocardio, esteatosis macro y microvesicular en el hígado y congestión multivisceral.
Los cultivos post mórtem fueron negativos en todos los órganos, a excepción del colon, donde se desarrolló Kleibsiella pneumonaie. Sin embargo, no existieron datos histológicos de infección.
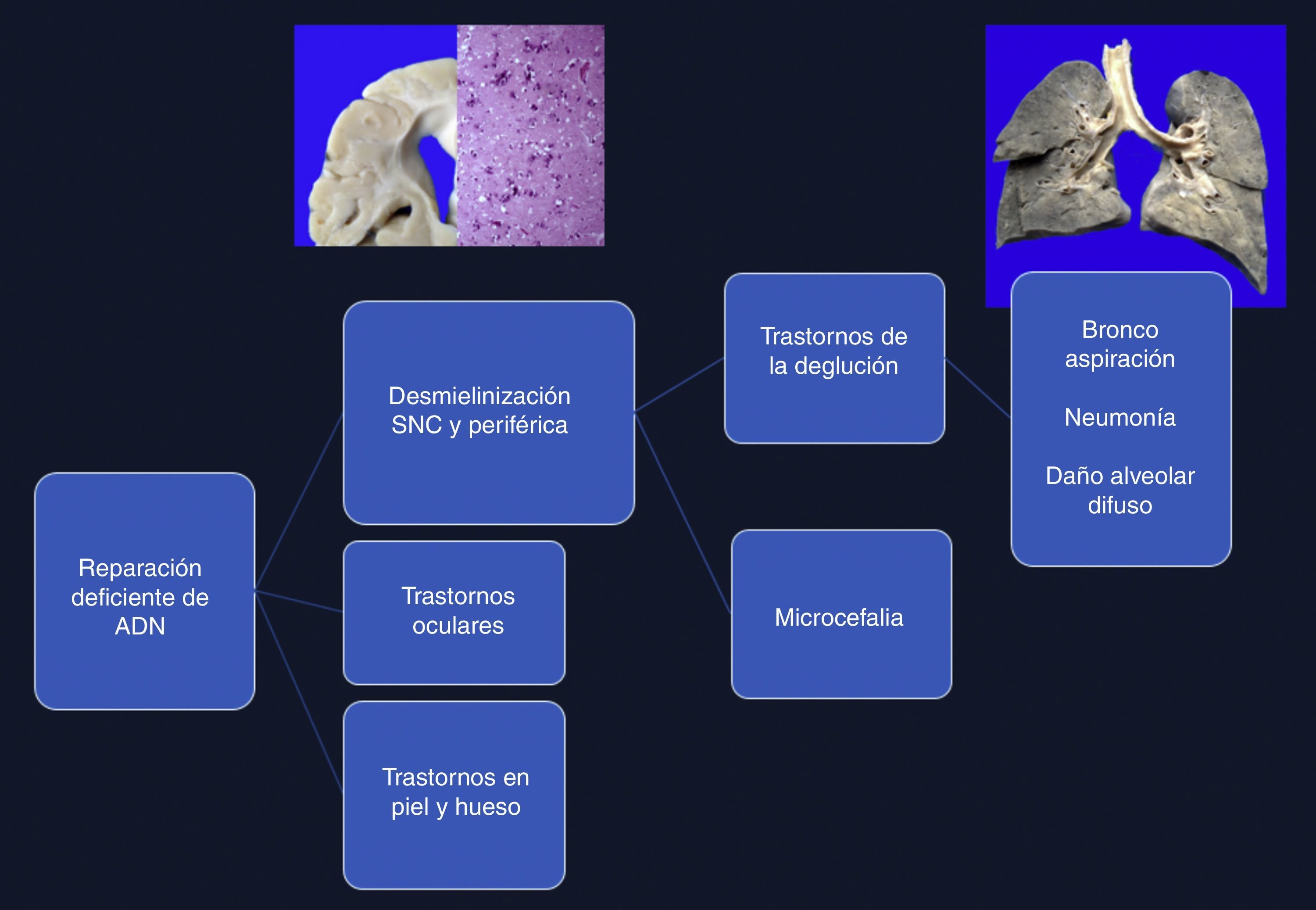
4Comentarios finalesEl síndrome de Cockayne está asociado con una mutación en los genes ERCC6 (5q11), en el 75% de los casos, y ERCC8 o CKN2 (10q12), en el 25% de los casos. Ambos codifican para las proteínas CS-A y CS-B, que están relacionadas con la reparación del ADN1–3. Las alteraciones ocasionan la acumulación de ADN dañado, y por consiguiente el envejecimiento celular y alteraciones en el crecimiento celular. La causa de la muerte en esta paciente se explica por una desmielinización en sistema nervioso central y periférico, que produjo un trastorno en la deglución, lo cual condicionó una broncoaspiración crónica de larga evolución, múltiples episodios de inflamación en los pulmones, y finalmente daño alveolar difuso extenso que la llevó a la muerte. Las alteraciones en la reparación de ADN ocasionaron trastornos oculares, en la piel y en los huesos (Figura 12)4–6.

Existe una gran variedad de causas que ocasionan catarata congénita. Antes de considerar alguna entidad genética, es necesario descartar infecciones maternas durante el embarazo. También hay que investigar errores innatos del metabolismo, como galactosemia, que ocasionalmente pueden presentarse con catarata desde el nacimiento. Es importante realizar una exploración clínica minuciosa para diferenciar una catarata congénita aislada de una catarata congénita sindrómica. Dentro de las sindrómicas existen, a su vez, dos grandes grupos: las monogénicas y las cromosomopatías.
4.2Oftalmología (Dra. Tania Valderrama)Esta paciente llegó con una catarata total bilateral, por lo que requería un abordaje multidisciplinario entre pediatría y genética. Se realizó la cirugía del ojo izquierdo y después la del derecho. No se colocó lente intraocular ya que estos pacientes con enfermedad sistémica cursan con procesos inflamatorios más intensos y se tiene el riesgo de complicaciones como glaucoma. La catarata bilateral era total, con metaplasia de la cápsula posterior, lo cual es frecuente en pacientes con síndromes sistémicos. Además de la catarata congénita, los niños con síndrome de Cockayne pueden tener enoftalmos, como en esta paciente, distrofias retinianas, estrabismo y nistagmus por mala visión.
Lamentablemente, en México los resultados de las cirugías en este tipo de pacientes son malos, ya que llegan en estadios muy avanzados. Lo ideal es someterlos a cirugía antes de los tres meses de vida para que puedan tener una mayor calidad de visión.
4.3Gastronutrición (Dr. Salvador Villalpando)Ante el hallazgo de la desnutrición severa y el cuestionamiento sobre el momento oportuno de formas alternativas de nutrición enteral, se comentó la importancia de tomar en cuenta el pronóstico de vida del paciente y evitar el encarnizamiento terapéutico con medidas invasivas.
4.4Cirugía (Dr. Ricardo Ordorica)En esta paciente se tomó la decisión de realizar la funduplicatura y la gastrostomía basados en la frecuencia y número de eventos de broncoaspiración que había presentado y la mala la mecánica de la deglución. En nuestra casuística de 96 casos, el 75% de los pacientes a los que se les realizó funduplicatura tenían daño neurológico. Si bien este procedimiento tiene complicaciones, como la estenosis esofágica y el vaciamiento acelerado, mejora el estado nutricional de los pacientes y su calidad de vida.
4.5Pediatría (Dra. Saskia Bermúdez)Esta es una enfermedad progresiva, con evolución clínica desfavorable y asociada con múltiples comorbilidades. La sobrevida generalmente es menor a los 10 años de edad. Se tiene la responsabilidad de brindar a los padres de este tipo de pacientes asesoramiento genético, así como información sobre el pronóstico fatal. Es importante discutir el caso con un comité de ética.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.