









Introducción: La incontinencia pigmenti es una enfermedad genética rara ligada al cromosoma X, letal en el varón, que afecta a todos los tejidos derivados del ectodermo, como piel, faneras, ojos, dientes y sistema nervioso central, y presenta alteraciones de grado variable en la inmunidad celular. Se caracteriza por la disminución de la melanina en la epidermis y su incremento en la dermis.
Caso clínico: Se presenta el caso de una lactante de dos meses de edad con incontinencia pigmenti grave, confirmada con estudio histopatológico de piel, que cursó con alteraciones neurológicas severas y crisis convulsivas. Además, presentó inmunodeficiencia celular grave que condicionó el desarrollo de infecciones que le ocasionaron la muerte.
Conclusiones: Se resalta la importancia del diagnóstico clínico temprano, así como la importancia del manejo multidisciplinario de las alteraciones neurológicas y de las complicaciones infecciosas.
Background: Incontinentia pigmenti (IP) is a rare, X-linked genetic disease and affects all ectoderm-derived tissues such as skin, appendages, eyes, teeth and central nervous system as well as disorders of varying degree of cellular immunity characterized by decreasing melanin in the epidermis and increase in the dermis. When the condition occurs in males, it is usually lethal.
Case report: We present the case of a 2-month-old female infant with severe IP confirmed by histological examination of skin biopsy. The condition evolved with severe neurological disorders and seizures along with severe cellular immune deficiency, which affected the development of severe infections and caused the death of the patient.
Conclusions: The importance of early clinical diagnosis is highlighted along with the importance of multidisciplinary management of neurological disorders and infectious complications.
1. Introduction
Incontinentia pigmenti (IP) or Bloch-Sulzberger syndrome is a rare multisystem X-linked genetic disease. The disease is often lethal in males and affects all ectoderm-derived tissues such as skin, appendages, eyes, teeth and central nervous system, as well as alterations of variable degree in cellular immunity. It is named for the reduction or absence of melanin in the basal cells of the epidermis and its increase in the dermis; therefore, the lesions adopt a whitish appearance. Histopathological finding of the third stage of the disease is precisely incontinence or pigment release to the dermis.1 The first description of IP was made in England in 1906 by Garrod. However, the disease was clinically characterized in 1920 by Bardach, Bloch, Siemens and Sulzberger, although only the names of Bloch and Sulzberger appear in the eponym of the disease.
The majority of IP cases appear sporadically as a result of a de novo mutation consistent with the deletion of a large part of the gene NEMO (nuclear factor kappa B essential modifier [NF-κB essential modulator] or IKKγ [IκB kinase-γ]) that includes axons 4-10 and is found on loci Xq28. This gene controls the expression of some cytokines that protect the cell against the TNFα-induced apoptosis.2,3 The condition is often lethal in males who inherit the mutated gene. Few cases of males who have survived have been reported and have been related with the presence of an extra X chromo-some or with somatic mosaicism. Affected females survive due to the presence of two X chromosomes (dizygocity of chromosome X) and to the early inactivation of chromosome X produced by the mutation.4,5 Recently, IP has been related to the presence of hypomorphic alleles, i.e., genetic mutations that do not completely nullify the function of protein coding. These hypomorphic mutations result in an allelic variant of IP called hypohydrotic ectodermic dysplasia associated with severe immunodeficiency (ADE-ID). The immunodeficiency presented by these patients could be hypogammaglobulinemia with low levels of IgG, hypergammaglobulinemia IgM (hyper-IgM phenotype) or dysgammaglobulinemia due to the mutation of the NEMO gene generating a deficient CD40 pathway signaling, a molecule that activates lymphocytes causing a faulty response to polysaccharide antigens and a high susceptibility to serious infections. It has also been described that NEMO participates in the signaling pathway for cellular cytotoxicity by the NK, which can cause recurrent viral infections.6-8
Morbidity due to IP is reported to be 0.7/100,000 with a frequency of presentation of ~1/50,000 live newborns and only accounts for 1% of all neuroectodermal disorders. In Mexico, scarce data are available on the incidence of IP. In a review of the National Institute of Pediatrics, 36 patients were diagnosed in 20 years, which accounts for ~1.8 new cases per year.9,10
Although IP is a systemic disease, the principal and always constant clinical manifestation is the skin condition, which usually courses through four evolutionary stages:
1. Erythematous, vesicular and occasionally pustular lesions especially in the extremities and trunk that generally appear from birth or during the first weeks of life, although in some cases these have developed in utero.
2. Verrucous, keratotic lesions between the 2nd and 6th weeks of life that appear on trunk, limbs and toes. They can persist for months and disappear without sequelae.
3. Hyperpigmented lesions that usually appear from the 4th month of life following the Blaschko lines. This tends to be the most persistent stage and is accompanied by alopecia. Lesions may disappear during the 2nd or 3rd decades of life.
4. Whitish hypomelanic striped macules on the legs that may be the only manifestation of IP in the adult. During this last stage there may also be skin atrophy.11-17
The clinical expression of IP is very variable even in members of the same family affected. Some patients have only skin and dental alterations, whereas others may have a severe and progressive course with fatal infections. Others may develop severe neurological and ophthalmological disabilities.
Clinical diagnosis of IP can be established according to the criteria proposed by Landy and Donnai12 (Table 1); 50-80% of cases are accompanied by extradermal alterations. The majority of patients have dental dysplasias such as hypodontia, anodontia, delayed eruption, crowns in the shape of a cone, supernumerary cuspid, enamel anomalies and retained teeth; 40% of patients have ocular disorders such as strabismus, retinal hypopigmentation, proliferative retinopathy, cataracts and corneal spots. Neurological disorders are seen in more than half of the cases, but only 7.5% present with severe neurological manifestations; 40% present with seizures manifested from the first week of life. Neuroimaging studies with nuclear magnetic resonance may show periventricular leukomalacia, hypoplasia of the corpus callosum, heterotopia of the gray matter, brain anomalies, encephalomalacia, brain infarcts and vascular anomalies.

Occasionally there may be demyelinating lesions without clinical impact. It has been suggested that ophthalmologic and neurological alterations are secondary to a microangiopathic process with secondary ischemia at a time of high vulnerability of the newborn and that the eosinophilic degranulation secondary to the activation of eotaxin causes direct tissue damage. Therefore, changes seen on the retinal vasculature could serve as a potential marker of central nervous system diseases. Other changes described in patients with IP are mammary hypoplasia, endomyocardial fibrosis, tricuspid insufficiency and pulmonary hypertension.18-26
With respect to alterations in immunity, it is known that NEMO (NF-κB essential modulator) is an intracellular molecule involved in cellular activation by the NF-κB pathway, which is activated during the inflammatory response by certain cytokines such as TNF-α and IL-1β and by different products of microorganisms that recognize innate immunity receptors.
The NEMO gene is involved in the development and function of the immune system. It is an intracellular molecule that responds rapidly before a large variety of stimuli involved both in the innate immune response as well as the adaptive, such as the Toll type receptors, receptors that have to do with programmed cell death or by damage (NOD/ CARD), receptors of the IL-1 and TNF superfamily, IL-15 receptor, IL-17 receptor, and T- and B-cell receptors. Activation of NEMO will produce the expression of a variety of genes involved in immunity such as acute phase proteins, cytokines, growth factors, adhesion and co-stimulation of surface molecules and regulators of cell proliferation and apoptosis.
The manifestations described classically in patients with immunodeficiency and NEMO mutations are serious bacterial infections of the airways, skin, soft tissue, bone, gastrointestinal tract, central nervous system and sepsis, the causal agents being Gram-positive bacteria such as Streptococcus pneumoniae and Staphylococcus aureus followed by Gram-negative bacteria, mainly Pseudomonas spp. and Haemophilus influenzae, in addition to mycobacteria.
NEMO mutations (E315a and R319q) associated with the Mendelian syndrome of susceptibility to mycobacterial infections (MSMD) have a phenotype where the microbiodependent pathway is found to be intact. Blood of these patients is normal where it is activated for BCG and cytokines such as IL-12 and IFN-γ. In turn, with PHA-activated mononuclear cells or CD3 T-dependent pathway, there is a defect in the production of IL-12 and secondarily of IFN-γ. In cell cultures of monocytes and T-lymphoctyes activated by PHA or CD3, defect in monocytes is evident as they cannot produce IL-12.27,28 These NEMO mutations produce a signaling deficiency in the activation by the CD40-CD40L pathway because according to the model of the NEMO protein, the mutations would produce a break in the saline bridge and interact with the CD40 pathway. The response to the drug or combined treatment with IFN-γ in patients with these mutations in NEMO is variable and patients commonly have mycobacterial infections.
Due to the mutation referenced in the NEMO gene, the affected cells are refractory to NF-kB-dependent enhancement and are therefore highly susceptible to TNF-induced apoptosis, suggesting that this cytokine is mainly involved in the appearance of lesions in IP and is also associated with other anomalies in the immune response.29-32
Laboratory blood studies reported leukocytosis and eosinophilia in 5-79% of the patients. The highest levels are observed between the third and fifth week of life, coinciding with the vesicular phase.33-37 Histopathological study of the skin lesions confirms the diagnosis where the histological findings will depend on the stage during which the biopsy is performed.
• In the first phase there are intraepidermal vesicles, eosinophilic spongiosis, single dyskeratotic cells and swirls of squamous cells with central keratinization. There is inflammatory infiltrate in the dermis with predominance of eosinophils and rare mononuclear cells.
• In the second phase, hyperkeratosis, acanthosis and papillomatosis are observed. Intraepidermal keratinization (evidenced by the swirls of squamous cells with central keratinization and dyskeratosis) is more pronounced than during the inflammatory phase. There is scant chronic inflammatory infiltrate and some melanophages are seen in the dermis.
• In the third stage of the disease, histopathology showed normal epidermis or slightly acanthotic and abundant macrophages loaded with melanin or melanophages are seen in the papillary dermis.
• In the fourth stage we observe a decrease of pigmentation of the basal layer and in the dermis the skin annexes are atrophic or absent.38,39
2. Clinical case
We present the case of a 3-month-old female infant of 37-year-old non-consanguineous parents. However, both parents came from a small community and shared the same last name, so there was the possibility of inbreeding. One sister died at 2 months of age due to probable sepsis. There were three healthy sisters. The newborn was the product of a fifth pregnancy without any abnormal characteristics and the infant was born via vaginal delivery at 38 weeks of gestation. Birth weight was 3400 g, length 50 cm, and Apgar score was unknown, but the mother reported that the infant cried and breathed at birth. Infant and mother were discharged 24 h after delivery.
At 8 days of life the patient presented fever, runny nose and cough. She was brought to the local hospital where infection of the upper respiratory tract infection and facial erythema were diagnosed. At 15 days she presented with hyperpigmentation on the dorsum of the nose that extended to cheeks, chest, hands and feet.
At 18 days of life she presented pneumonia with respiratory failure and was admitted to a local hospital with sepsis, necrotizing enterocolitis, seizures, anemia and thrombocytopenia. Due to poor clinical evolution and persistence of fever, she was transferred to the Hospital Infantil de Mexico Federico Gomez in Mexico City.
At the time of her admission she was in poor general condition. On physical examination there was tissue hypoperfusion. The skin was hard and of cardboard consistency, showing generalized hyperpigmentation predominantly in the face and chest and genitalia without signs of virilization or macro-genitosomia. There was spasticity of the extremities (Figure 1).
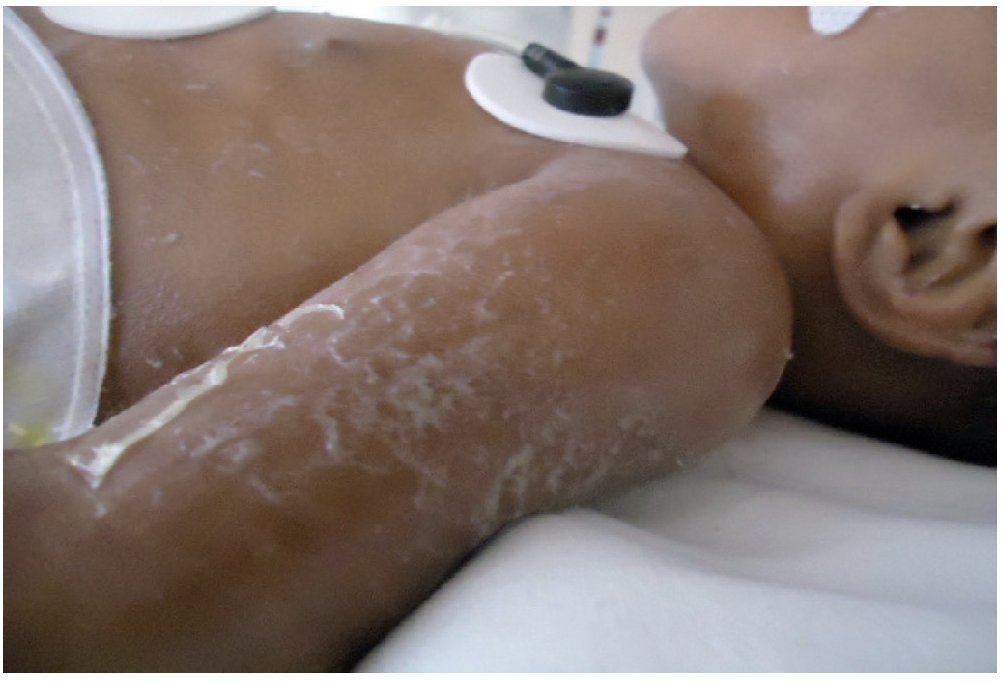
Figure 1 Hiperpigmentation in face, trunk, and extremities with scaly formation.
Initial laboratory examinations showed hemoglobin 7.6 g/dl, leukocytes 40,600/mm3, neutrophils 70%, bands 3%, lymphocytes 2%, monocytes 24%, eosinophils 0%, basophils 0%, platelets 25,000/mm3, glucose 3 mg/dl, BUN 10 mg/dl, creatinine 0.4 mg/dl, uric acid 3.6 mg/dl, sodium 122 mEq/l, potassium 2.9 mEq/l, chloride 86 mEq/l, calcium 8.0 mg/dl, phosphorus 3.3 mg/dl, total bilirubin 1.56 mg/dl, direct bili-rubin 0.34 mg/l, albumin 3.4 g/dl, ALT 28 U, AST 61 U, GGT 71 U, lactose dehydrogenase 592 U, alkaline phosphatase 136 U, PT 12.8”, PTT 44.7”, INR 0.99, and fibrinogen 259 mg/dl.
Arterial blood gas showed pH 7.51, pCO2 47.1, pO2 131, HCO3 37.4, and lactate 1 mmol/l. The patient was initially managed according to the protocol of nosocomial sepsis with pneumonia with the administration of i.v. fluids, ventilatory and inotropic support, and a double scheme of antibiotics with cefepime and amikacin. Also, due to skin hyperpigmentation with shock and hyponatremia she was managed as an adrenal crisis, probably secondary to congenital adrenal hyperplasia. However, ACTH levels of 25.3 pg/ml, cortisol 13.4 μg/dl, androstenedione 312 pg/dl, testosterone <20 ng/dl, DHE-S 20.4 μg/dl were found to be within normal limits with the exception of cortisol, which was elevated due to the infectious process.
Lumbar puncture was done and cytochemical analysis of the cerebrospinal fluid (CSF) was normal. Initial blood culture was positive for Klebsiella pneumoniae with multiple sensitivities. Serological results for herpesvirus type I and II, CMV, HIV, rubeola and VDRL were negative.
Echocardiogram reported normal venous and pulmonary systems, AV and VA concordance, ejection fraction 72%, fractional shortening 37% and systolic pressure of the right ventricle 37 mmHg. Ophthalmological examination showed normal fundus of the eye and left corneal leukoma. EEG study showed generalized slow activity in delta polypoint range and intermixed sharp waves in the right hemisphere.
Skull CT showed increase of the subarachnoid space frontally. Cerebral parenchyma was demonstrated with adequate gray-white matter ratio without space-occupying lesions. Hyperintensity at the level of the basal ganglia and thalamus was seen, compatible with bleed or brain infarct. Nuclear magnetic resonance (NMR) of the skull with contrast showed hypointense images in the basal nuclei in T1 and T2 sequences. There was thinning of the corpus callosum in all its segments and generalized widening of the subarachnoid space.
Bone marrow aspirate reported decreased cellularity, absent megakaryocytes, promyelocytes 2%, myelocytes 15%, metamyelocytes 14%, bands 10%, segments 6%, eosinophils 0%, monocytes 2%, lymphocytes 45%, basophils 2%, normo-blasts 2% and blasts 2%. Serum immunoglobulins were IgA 35.1 mg/dl (6-58 mg/dl), IgG 671 mg/dl (270-780 mg/dl), IgM 26.2 mg/dl (12-87 mg/dl), and IgE 16.9 IU/ml (0-100 IU/ml). There was a low population of lymphocytes demonstrated by flow cytometry in peripheral blood (Table 2). The last blood count showed hemoglobin 9.3 g/dl, leukocytes 1,000/ mm3, neutrophils 34.4% (340), lymphocytes 48.9% (490), monocytes 0.16% (16), eosinophils 0.9% (10), basophils 0%, and platelets 45,000/mm3.

During the patient’s stay she had a maculopapular rash followed by formation of blisters and vesicles in the flexion folds of the extremities (Figure 2) which, when burst, formed verrucous plaques with hyperkeratosis and changes in pigmentation of the skin of the trunk. Alopecia in the frontal region up to the vertex was also observed (Figure 3) and was managed with moisturizers and emollient creams.

Figure 2 Blisters and desquamation.

Figure 3 Frontal alopecia.
Skin biopsy reported skin with hyperkeratosis and focal parakeratosis. The rest of the epidermis showed dyskeratosis, exocytosis of lymphocytes and neutrophils and formation of intraepidermal vesicles (Figure 4). Band infiltrate consisting of lymphocytes and melanophages with pigment was observed in the junction of the epidermal and papillary dermis (Figure 5). The reticular dermis and subcutaneous tissue showed no alterations.
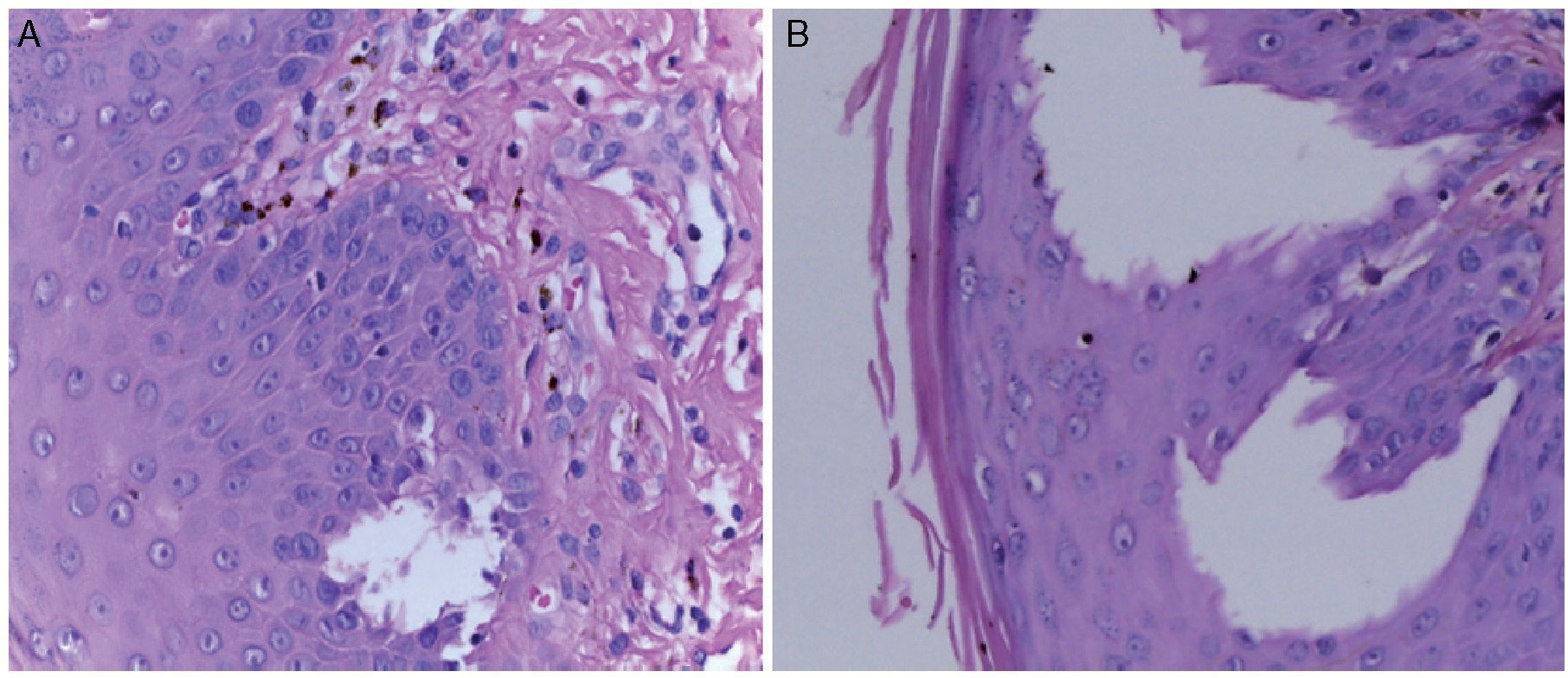
Figure 4 (A) Intraepidermal vesicle and output of pigment to the papillary dermis. (B) Intraepidermal vesicle.

Figure 5 Panoramic cut of the skin, dermal-epidermal junction with melanophages and output of pigment to the papillary dermis and changes in the stratum corneum of the epidermis.
Final diagnosis
Incontienentia pigmenti phase III.
Subsequently the patient had a torpid evolution. Hemo-dynamic and respiratory instability persisted as well as persistence of seizures, sepsis and amine-refractory septic shock. Antibiotic scheme was updated to meropenem, vankomycin and amphotericin B. However, despite intensive care and opportune updating of the antibiotic scheme, the patient developed multiple organ failure syndrome with renal insufficiency, acute liver failure, abdominal compartment syndrome, acute respiratory distress syndrome (ARDS) and disseminated intravascular coagulopathy (DIC). Refractory hypotension and metabolic acidosis persisted along with prolonged capillary refill, weak pulses and bradycardia, which led to irreversible cardiorespiratory arrest.
3. Discussion
This is the case of a female infant with a clinical picture compatible with severe IP, which initiated neonatally with skin, neurological and immunological involvement. Other causes of neonatal pemphigoid erythema were ruled out such as infections due to herpes simplex virus and congenital syphilis as well as other alterations of skin pigmentation such as hyperpigmentation secondary to congenital adrenal hyperplasia caused by the crossed reactivity of the adrenocorticotropin hormone (ACTH) with receptors for melanocyte-stimulating hormone (MSH)—and the benign transitory hyperpigmentation where there is no involvement of the skin appendages. Diagnosis of IP was confirmed with histo-pathological study of the skin biopsy.
This patient presented serious neurological condition related with anomaly of brain development and disorders in neuronal migration with hypoplasia of the corpus callosum along with vascular phenomena with ischemia and bleeding. This developed early, with difficult to control seizures accompanied by progressive neurological deterioration, which complicated her clinical condition. However, it should be mentioned that although in other cases of IP the common association of neurological changes with ophthalmological involvement has been reported, in this patient the only eye alteration detected was corneal leukoma.
It was considered that pulmonary arterial hypertension manifested by the increase of systolic pressure in the right ventricle was due to the severe lung condition secondary to pneumonia. However, this case illustrates how the diagnosis of this disease can be suspected in female infants who present to pediatric hospitals in poor general condition with sepsis and state of shock and who present at least one of the major diagnostic criteria for the disease such as vesicular exanthema, skin hyperpigmentation, Blaschko lines, verrucous plaques and frontal alopecia as well as the presence of some minor diagnostic criteria proposed by Landy and Donnai.12
No molecular studies were performed in this patient for determination of the type of NEMO gene mutation or tests for detecting possible delay in the inactivation of chromo-some X, which carries the mutation due to the severe clinical condition in which she arrived. No tests were carried out on the integrity and functioning of the interleukin-12 axis. Nevertheless, flow cytometry for counting of low population of lymphocytes showed severe lymphopenia [CD45 = 433, of which 99% were T lymphocytes (CD3 = 429) and only 1% were B lymphocytes] with immunoglobulins in the low normal limits. This caused the development of various severe infections during the patient’s short life as well as her death caused by a septic event refractory to adequate antibiotic treatment. This allows us to infer that the patient had severe alterations both of cellular as well as humoral immunity.
Based on the fact that the treatments proposed for this rare disease (IV gamma globulin and gamma-interferon) are expensive and inaccessible in our environment, it is believed that the possibility of offering timely treatment to IP patients will depend on establishing an early confirmatory diagnosis. This is in addition to the vital support in intensive care units of the seriously ill and unstable patient as well as the multidisciplinary approach and treatment of the infectious, neurological and ophthalmological changes.40-46 Finally, offering parents genetic counseling is essential.47-50
Ethical disclosure
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of data. The authors declare that no patient data appears in this article.
Right to privacy and informed consent. The authors declare that no patient data appears in this article.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 27 June 2015;
accepted 13 August 2015
☆ Please cite this article as: Zamora-Chávez A, Escobar-Sánchez A, Sadowinski-Pine S, Saucedo-Ramírez OJ, Delgado-Barrera P, Enríquez-Quiñones CG. Incontinencia pigmenti con defecto en la inmunidad celular. Bol Med Hosp Infant Mex. 2015. http://dx.doi.org/10.1016/j.bmhimx.2015.08.003
* Corresponding author.
E-mail:azamora@himfg.edu.mx (A. Zamora-Chávez).