We studied the role of Thermus thermophilus Recombinase A (RecA) in enhancing the PCR signals of DNA viruses such as Hepatitis B virus (HBV). The RecA gene of a thermophilic eubacterial strain, T. thermophilus, was cloned and hyperexpressed in Escherichia coli. The recombinant RecA protein was purified using a single heat treatment step without the use of any chromatography steps, and the purified protein (>95%) was found to be active. The purified RecA could enhance the polymerase chain reaction (PCR) signals of HBV and improve the detection limit of the HBV diagnosis by real time PCR. The yield of recombinant RecA was ∼35mg/L, the highest yield reported for a recombinant RecA to date. RecA can be successfully employed to enhance detection sensitivity for the diagnosis of DNA viruses such as HBV, and this methodology could be particularly useful for clinical samples with HBV viral loads of less than 10IU/mL, which is interesting and novel.
The recombinase A (RecA) protein (accession no. AAK15321.1) from the thermophilic bacterium Thermus thermophilus is a thermostable enzyme that plays important roles in homologous recombination and DNA repair.1 DNA recombination in prokaryotes is achieved by catalyzing the pairing of homologous DNA sequences,2,3 and DNA repair is achieved by protection of the damaged DNA ends.4,5 This protein has the activities of single-stranded DNA dependent ATPase, DNA annealing, and exchanging of strands between two recombining DNA double helices, similar to Escherichia coli RecA (accession no. AFU91764.1) protein, but the optimal temperature for the strand exchange of RecA is between 50 and 60°C.6
RecA-like proteins have been sequenced from over 60 different prokaryotic species7,8 and it hydrolyzes ATP when bound to DNA, and its ATPase activity rate is proportional to the amount of RecA bound to the DNA9–12 with the fact that the ATPase activity is used to measure its binding efficiency to DNA.13 It has been shown that the RecA protein catalyzes in vitro DNA strand exchange reactions14 with peak efficiency at approximately 60°C,6 a temperature that is fairly close to the Tm required for thermal denaturation of dsDNA.15
Numerous applications of RecA are reported. These include identification of different Burkholderia cepacia genomovars,16 sequence-specific ligation of DNA using the E. coli RecA protein,17 maintaining the integrity of chloroplast DNA molecules in Arabidopsis,18 auto-cleavage of other serine proteases and repressors of the bacteriophage lytic cycle,19–22 and its role in swarming motility in E. coli.23
Shigemori et al.24 have used RecA in stimulating pairing between completely matched primers and target sequences and effectively eliminating non-specific PCR products. This specificity enabled them to amplify the sequences between any sites of interest without needing to consider the optimal priming sites for each PCR. Similarly, Fukui and Kuramitsu25 have recently utilized MutS, a thermostable mismatch-recognizing protein from T. thermophilus, along with RecA to reduce non-specific amplifications in PCRs.
Hence, we realized the immense potential of RecA in PCR-based diagnostics and attempted to generate RecA protein by a simple purification method that would in turn make the PCR based detection of viruses more sensitive and cost-effective.
Material and methodsReagentsAll chemicals were of analytical grade. Oligonucleotides, used in this study, were from BioServe Technologies Pvt. Ltd, Hyderabad, India. Bradford protein assay reagents and IPTG were obtained from Amresco, USA. Standard RecA pure protein was procured from NEB, USA, while ATP was purchased from Thermo Fisher Scientific, USA. Protein molecular weight markers 11–180kDa and 11–97kDa were obtained from G-Biosciences, USA and Aristogene Life Sciences, Bangalore, India respectively.
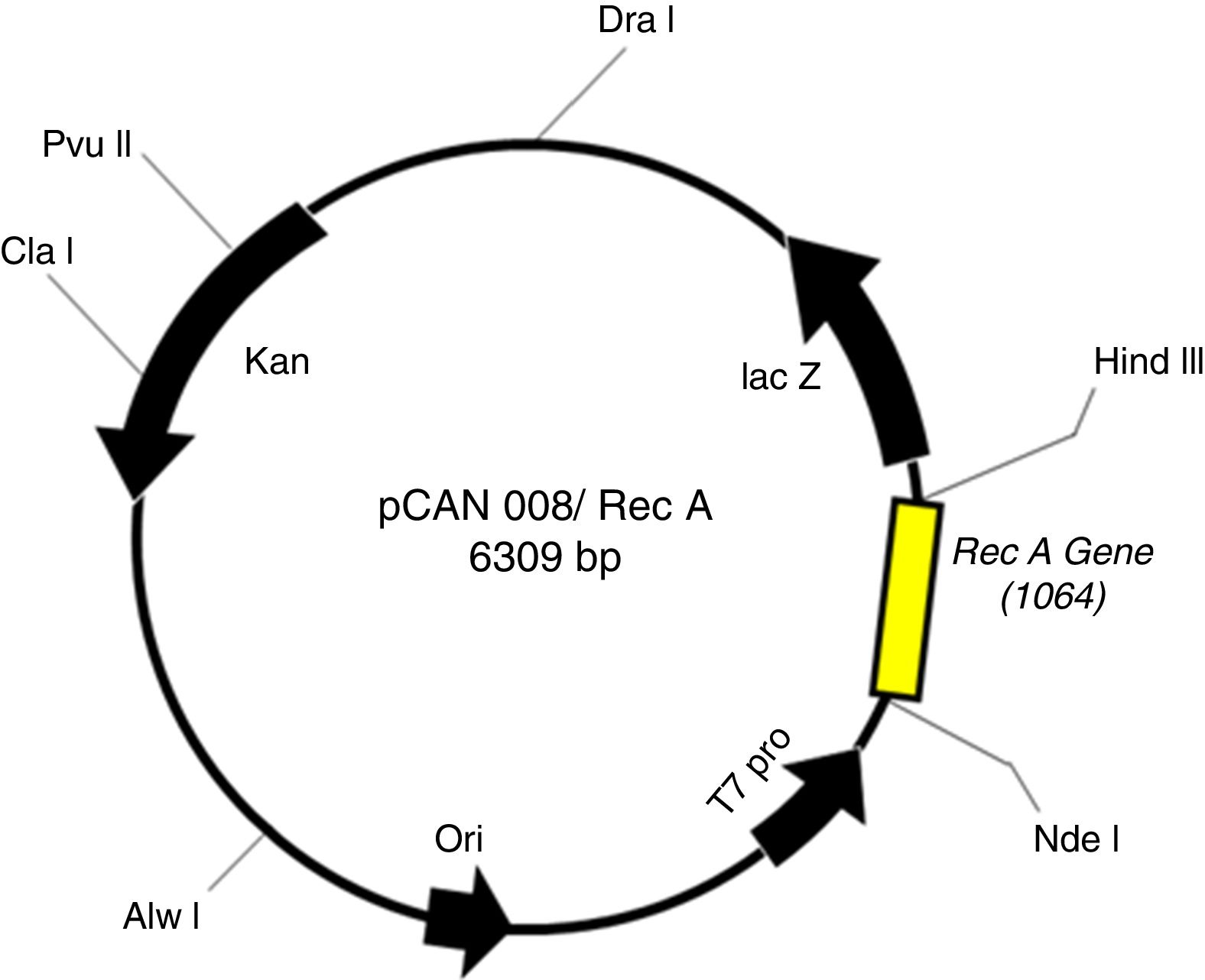
Cloning and construction of RecA expression constructThe synthetic gene of T. thermophilus RecA was custom synthesized from Integrated DNA Technologies, Inc. (IDT), USA, and cloned into the pUC57 vector. The nucleotide sequence of the T. thermophilus RecA gene was adjusted for codon bias without altering any of the amino acids. The RecA gene was excised from the pUC57-RecA plasmid as a NdeI/HindIII fragment and cloned into the pET26b vector into similar restriction sites. The recombinants were screened by PCR using T7 promoter specific primers, forward 5′ TAATACGACTCACTATAGGG 3′ and reverse 5′ GCTAGTTATTGCTCAGCGG 3′, and the positive clones were further confirmed by restriction analysis and DNA sequencing. The pET26b-RecA plasmid was designated as pCAN008.
The plasmid DNA of pCAN008 was introduced into ER2566 competent cells (NEB, USA) for expression of recombinant RecA. The protocol was essentially as described by Paul et al.26 Briefly, the induction of RecA was effected by the addition of 1mM IPTG when cells in LB medium (containing 20μg/mL kanamycin) reached an OD600 value between 0.5 and 0.8. The induction proceeded for 4h by incubating the culture at 37°C, and then the induced cells were pelleted at 7670×g for 10min. The harvested cells were resuspended in 25mM Tris. Cl, pH 7.5 and sonicated in a sonicator (Omni Sonic Ruptor 400, GA, USA) at 50% amplitude for one min on/off for 10min in cold. The sonicated lysate was spun at 9000×g for 20min at 4°C and the supernatant obtained was used for purification of the RecA expressed protein.
RecA purification by heat treatmentRecA is known to be thermostable,27 and we utilized this property to purify the recombinant RecA protein. Different volumes of crude lysate of RecA were heated at 75°C for 30min, after which the samples were centrifuged at 30,670×g for 10min, and the supernatants were analyzed on 12% SDS–PAGE for RecA protein followed by silver staining as described by Nesterenko et al.28 Protein concentrations were determined by Bradford's method29 to quantify the total yield of RecA per liter of induced culture under the described experimental conditions.
ATP hydrolysis assayATPase activity was determined using a QuantiChrom ATPase/GTPase Assay Kit DATG-200 (EnzyChrom, USA). Phosphate standards were made by taking varying concentrations of phosphate (0–50μM) in nuclease free water as per manufacturer's instructions. Equal amounts of proteins of in-house purified RecA and commercial RecA (New England Biolabs, UK) were used for detecting the ATPase activity. After incubating the RecA proteins with ATP, 200μL of the supplied reagent was added, and contents were incubated for 30min at room temperature. The OD of the samples, reflecting free phosphate release due to ATP hydrolysis by RecA, was read at OD620nm in an ELISA plate reader (Tecan Group Ltd, Männedorf, Switzerland).
Human leucocyte antigen (HLA) PCRHuman gDNA is the source of the template for the amplification of HLA genes.30 The different protocols for HLA typing reported require use of gDNA between 5 and 30ng/μL31 and DNA concentrations lower than 2ng/μL have been found to compromise genotyping results severely.32 Since RecA is known to prevent false priming, we hypothesized that samples with lower concentrations of genomic DNA might display higher gene amplifications in presence of RecA. To validate this hypothesis, we carried out PCRs of HLA genes from human gDNA in presence and absence of RecA. The primers for amplification of HLA class I genes, namely, HLA-A-exon 2 and exon 3, HLA-B-exon 2 and exon 3 and HLA-C-exon 2 and 3, were as described by Itoh et al.,33 while the primers for HLA class II genes such as DPB1, DQB1 and DRB1, targeting exon 2, were according to Lange et al.30 The PCR reaction was carried out in 25μL reaction volume and the PCR mixture comprised 1× ammonium sulfate buffer, pH 8.3; 2.5mM magnesium chloride; 0.2mM dNTPs; 5% DMSO; 0.2% Tween-20; 0.04% BSA; 0.4μM HLA primers; and 1unit of Taq DNA polymerase (ThermoFisher Scientific, USA) with 40ng of gDNA. PCR conditions were as follows: Initial denaturation of 95°C for 5min, followed by 35 cycles of 95°C, 30s; 51.1°C, 30s; and 72°C, 45s. The final extension step was performed at 72°C for 10min, and suitable aliquots were loaded on a 2% agarose gel for visualization.
Sample collection and HIV, HBV and HCV PCRPatients who visited Kuppam Medical College, Andhra Pradesh, India for viral testing of HIV, HBV and HCV during the period of March 2016 to March 2017 served as the study material. Blood samples were collected in EDTA coated tubes and plasma was collected by centrifuging the samples at 2000×g for 10min. The plasma samples were stored at −20°C until further use. In some cases, serum was collected from the patients.
Plasma or serum samples were analyzed for HIV, HBV and HCV by routine diagnostic tests (RDT) as per manufacturer's instructions. The test kits employed included the HIV-1 TRI-DOT kit, HEPACARD test kit and HCV TRI-DOT test kits. Viral positive samples were handled with adequate precautions in the BSL-II facility of Cancyte Technologies Pvt. Ltd, Bangalore, India. IVD certified NucleospinDx virus kit (Macherey-Nagel, GmbH) was used to isolate viral nucleic acids following manufacturer's instructions.
To minimize the risk of contamination of PCR amplicons, and to avoid transfer of DNA from one sample to the other, in our laboratory, the pre and post-polymerase chain reaction (PCR) products were kept physically separated, and all biological samples were processed separately in a dedicated BSL-II biosafety hood. The DNA-free consumables and negative controls were used at every stage of the PCR analysis.
The HIV primers were designed to amplify the gag gene of the HIV (accession number KY713247.1), while the HBV and the HCV primers amplified the S gene (accession number MF967563.1) of HBV and 5′ UTR (accession number MG436884.1) region of HCV respectively. All the PCRs were carried out in 25μL reaction volumes and the PCR products were analyzed on 2% agarose gel electrophoresis stained with ethidium bromide for visualization, unless mentioned otherwise.
The PCR mixture comprised 1× Superscript III RT/Platinum® Taq Mix buffer, HIV forward primer (5′ ATCAAGCAGCCATGCAAAT 3′) and reverse primer (5′ TACTAGTAGTTCCTGCTATGTC 3′) or HCV forward (5′ GAA AGC GTC TAG CCA TGG CG 3′) and reverse primer (5′ ACG CCC AAA TGG CCG GGC ATA GA 3′), and 0.5μL of Super Script III enzyme. PCR was performed in a Proflex PCR machine (Applied Biosystems, USA). PCR conditions were as follows: one cycle of 50°C for 30min to convert RNA to cDNA, followed by a single step of denaturation at 95°C for 5min and 40 cycles of 95°C for 30s, 55°C for 30s, and 72°C for 1min followed by a final extension at 72°C for 10min.
PCR for HBV detection in clinical samples was carried out in 25μL reaction volumes. The PCR mixture comprised of 1× ammonium sulfate buffer, pH 8.3; 2.5mM magnesium chloride; 0.2mM dNTPs; 5% DMSO; primers 0.4μM (FP 5′ CCCCCACTGGCTGGGGCTTGGT 3′ and RP 5′ AGGACGTCCCGCGCAGGATC 3′); and 1unit of Taq DNA polymerase (ThermoFisher Scientific, USA). PCR steps included an initial denaturation of 95°C, 10min, followed by 40 cycles of 95°C, 30s; 60°C, 30s; and 72°C, 45s and a final extension step at 72°C for 10min.
For carrying out PCRs with RecA and ATP, ATP was used at a final concentration of 0.3mM along with 0.25μg of either in-house purified RecA or commercially available RecA.
HBV viral loadBriefly, the reaction was carried out in a final volume of 25μL containing 1× Thermo Scientific Maxima SYBR Green master mix, and forward (FP 5′ CCCCCACTGGCTGGGGCTTGGT 3′ and RP 5′ AGGACGTCCCGCGCAGGATC 3′) primers were added to a final concentration of 4pmol per reaction. A total of 0.3mM ATP and 0.25μg of RecA were added to one set, and suitable samples without RecA served as negative controls.
The qPCR was performed in a Qiagen qPCR machine (Qiagen, Germany), and the conditions for qPCR were 45 cycles of PCR amplification, each comprising 95°C for 5min, 95°C for 30s, 60°C for 30s, and 72°C for 30s. Data collection was performed at 72°C in each cycle. Following the amplification, a melting curve analysis was performed to verify the authenticity of the amplified products by checking their specific melting temperatures (Tm). Experiments were done in duplicates.
The recombinant plasmid containing the S gene of HBV (named pTZ57R/T-HBV) was confirmed by DNA sequencing (data not shown) and used to generate a standard curve. The concentration (ng/μL) of the purified stock of plasmid DNA was measured by a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific), and plasmid copy number was calculated using the DS DNA copy calculator using the formula: pTZ57R/Tcopies/μL=(plasmid concentration [ng/μL]×6.022×1023)/(nucleotide length×1×109×650), as described by Staroscik.34 The plasmid stock solution was diluted with DNase/RNase-free distilled water, 10-fold serial dilutions containing 2.5×105–250copies/μL were made, and small aliquots were stored at −20°C for future use in generating standard curves. The Ct values were tested for statistical significance by Student's t-test.
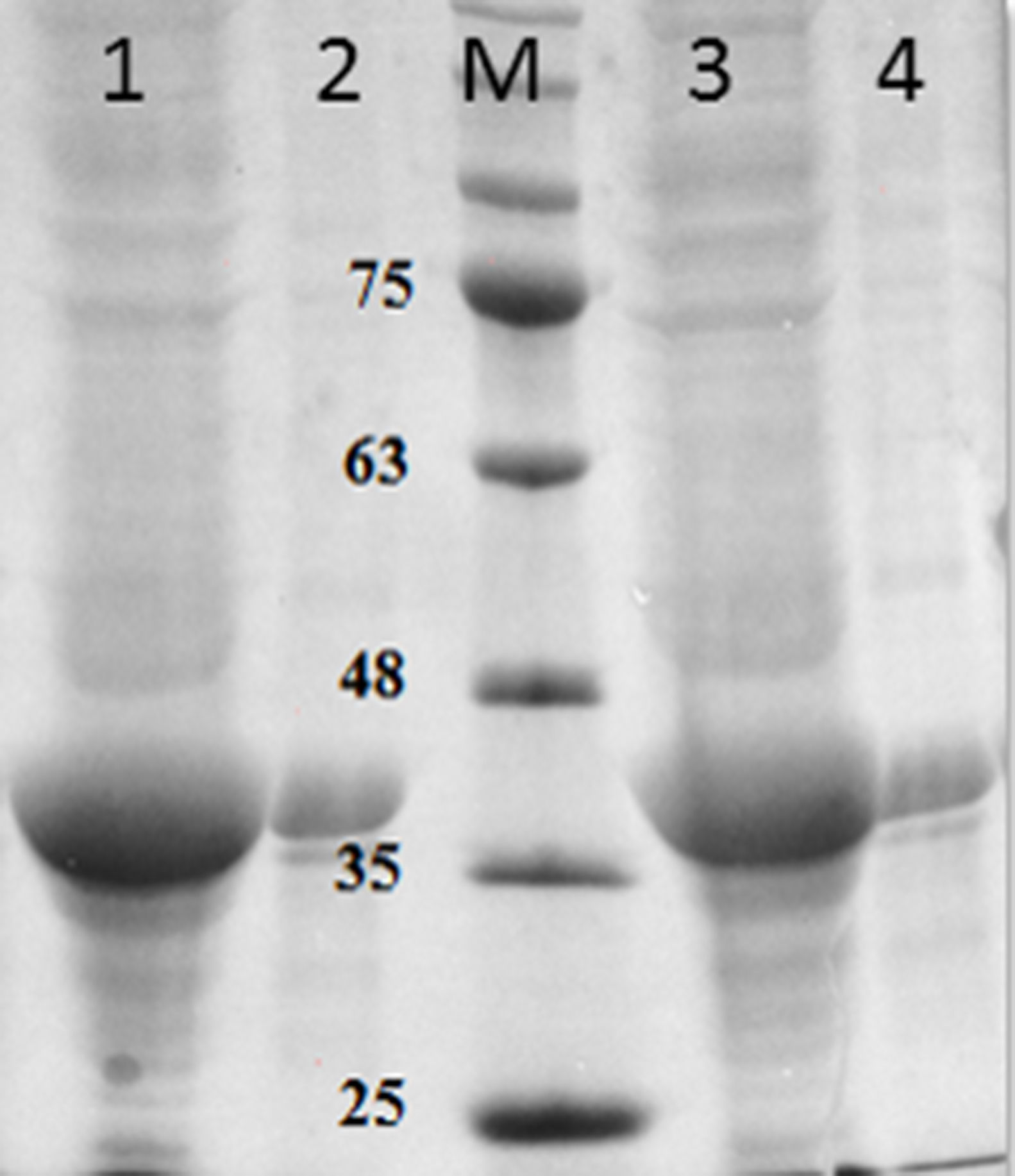
ResultsCloning and expression studiesFig. 1 shows the plasmid map of pET26b-RecA. The clones of pET26b-RecA were confirmed by DNA sequencing and restriction analysis. Fig. S1 shows the silver stained gel of whole cell lysates of two clones of pET26b-RecA. The recombinant RecA protein was restricted to the soluble fraction of the cell and constituted >50% of the total soluble protein as judged by densitometry scanning and of the expected size of ∼39kDa. The studies on the expression of RecA from pET26b-RecA clone in ER2566 cells using different concentrations of IPTG indicated that the amount of RecA expression increased with increased time of induction as expected (Fig. S2) and that the expression of RecA could be induced even with 0.125mM IPTG effectively (Fig. S3).
Purification and total yield of RecA using heat treatment.")
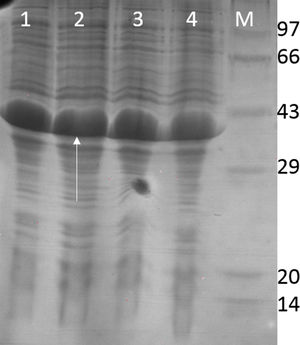
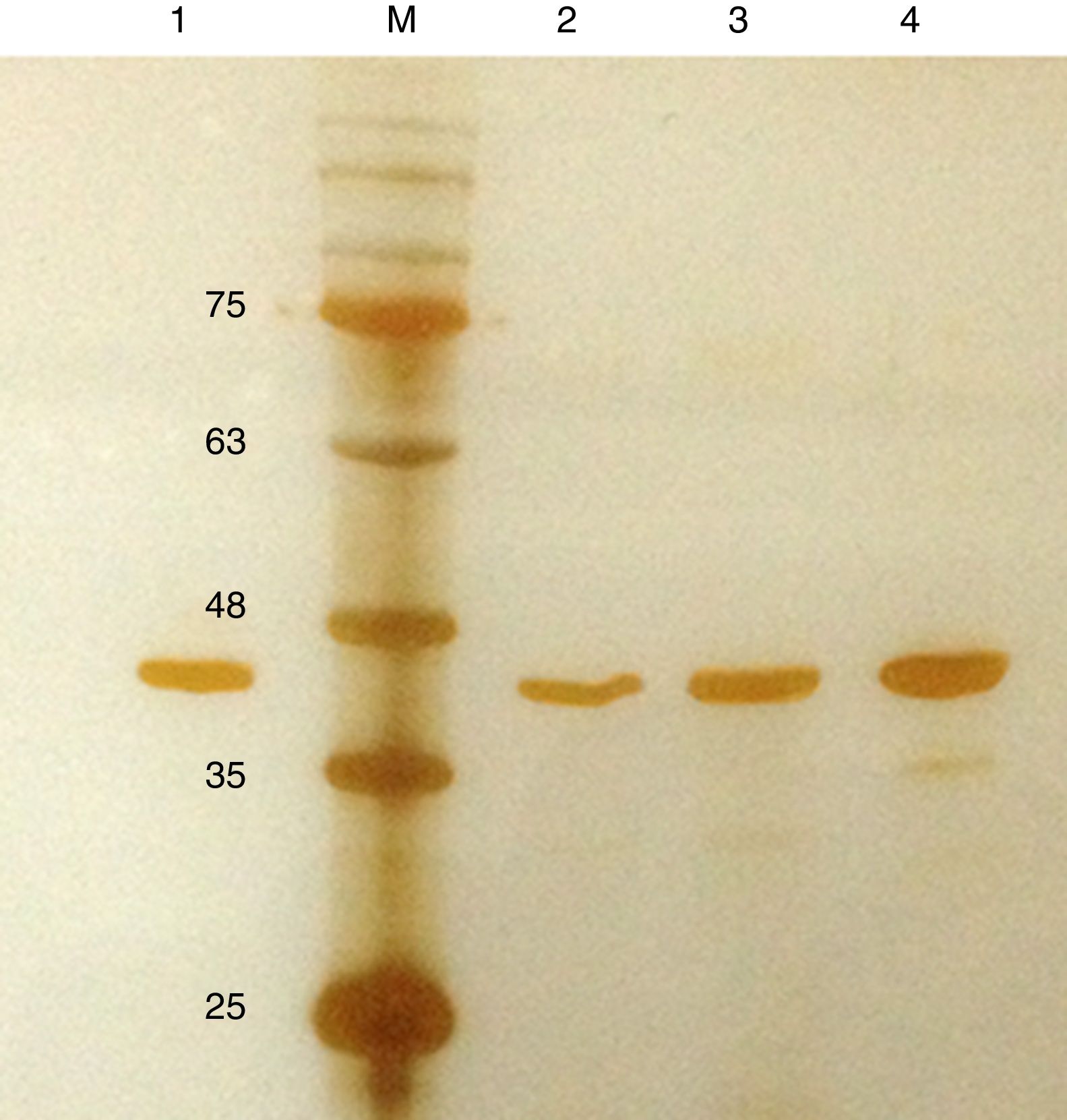
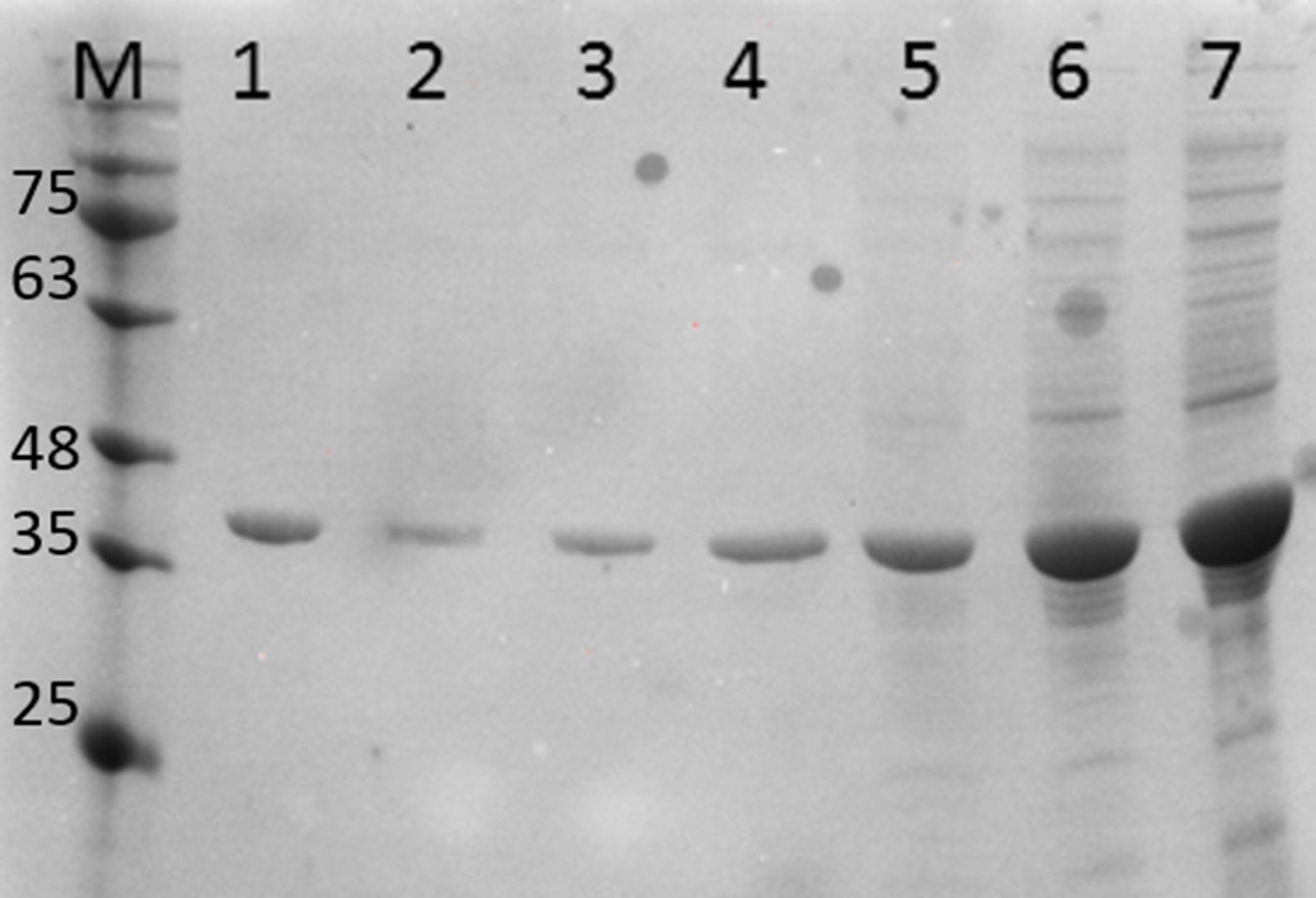
The RecA expressed from the clone pET26b-RecA (#5) after heat treatment was found to be homogenous and was >95% pure as judged by silver staining. It is evident from Fig. S4 that heat treatment was sufficient to achieve pure RecA. The total yield of pure RecA per liter of LB was found to be 35mg/L and its purity matched the purity of the commercially available RecA protein (Fig. 2).
; lane 2: in-house purified RecA (0.2μg); lane 3: in-house purified RecA (0.5μg); lane 4: in-house purified RecA (1.0μg). M: pre-stained protein MW marker (11–180kDa).")
Silver stained SDS–PAGE gel of the purified in-house RecA and commercial RecA proteins. Note that the purity of the RecA is >95% as judged by the silver stained gel. Lane 1: commercial RecA (0.25μg); lane 2: in-house purified RecA (0.2μg); lane 3: in-house purified RecA (0.5μg); lane 4: in-house purified RecA (1.0μg). M: pre-stained protein MW marker (11–180kDa).
The in-house purified RecA protein showed ATPase activity units of 3.0×105U/μL, while the commercial RecA showed activity of 2.5×105U/μL. This result indicated that the purification methodology followed for the in-house RecA protein does not compromise the function of the RecA protein.
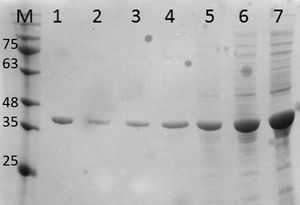
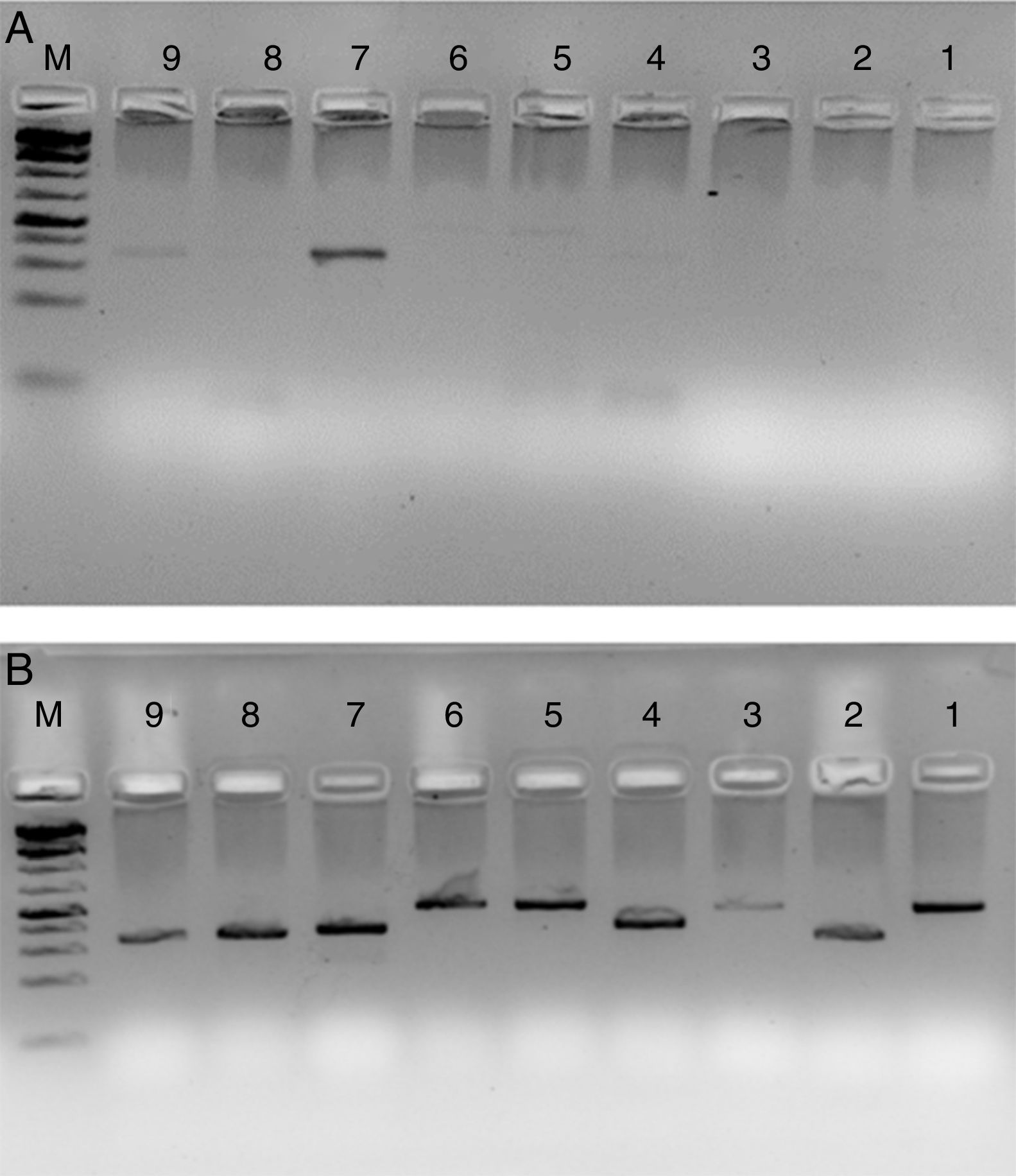
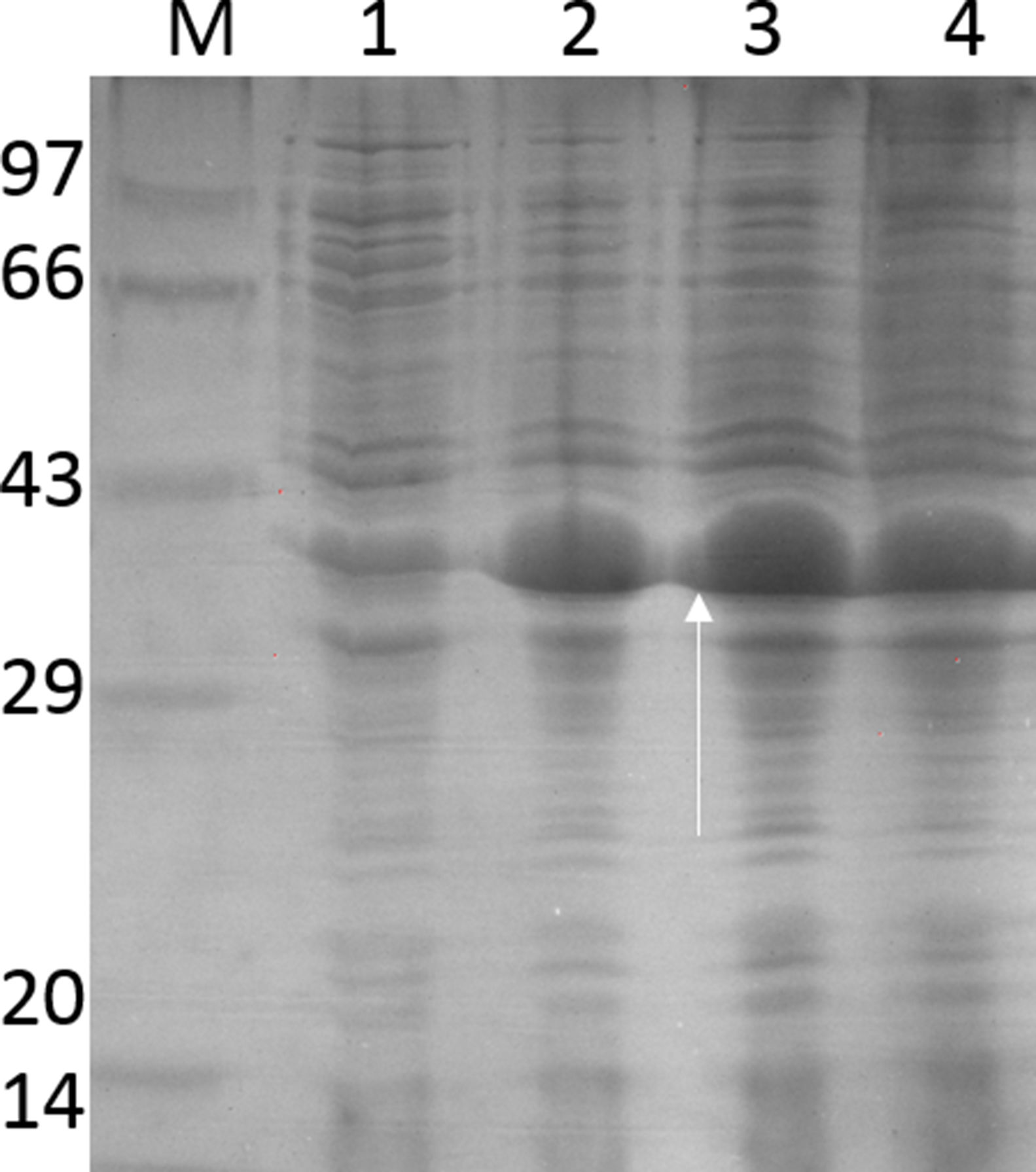
HLA-PCRIt is clear from Fig. 3 that RecA amplifies the HLA genes of interest even when the gDNA used was as low as 5ng per reaction, while without RecA, there was negligible amplification of all the genes under similar experimental conditions.
; lane 2: HLA-A-Exon 3 (327bp); lane 3: HLA-B-Exon 2 (469bp); lane 4: HLA-B-Exon 3 (371bp); lane 5: HLA-C-Exon 2 (489bp); lane 6: HLA-C-Exon 3 (500bp); lane 7: DPB1, Exon 3 (368bp); lane 8: DQB1, Exon 3 (350bp); lane 9: DRB1 Exon 3 (351bp); lane 10: DNA molecular weight marker (100bp ladder).")
HLA-PCR of Class I and Class II antigens by PCR. Panel A shows PCR without RecA, and panel B shows HLA PCRs with RecA. Lane 1: HLA-A-Exon 2 (478bp); lane 2: HLA-A-Exon 3 (327bp); lane 3: HLA-B-Exon 2 (469bp); lane 4: HLA-B-Exon 3 (371bp); lane 5: HLA-C-Exon 2 (489bp); lane 6: HLA-C-Exon 3 (500bp); lane 7: DPB1, Exon 3 (368bp); lane 8: DQB1, Exon 3 (350bp); lane 9: DRB1 Exon 3 (351bp); lane 10: DNA molecular weight marker (100bp ladder).
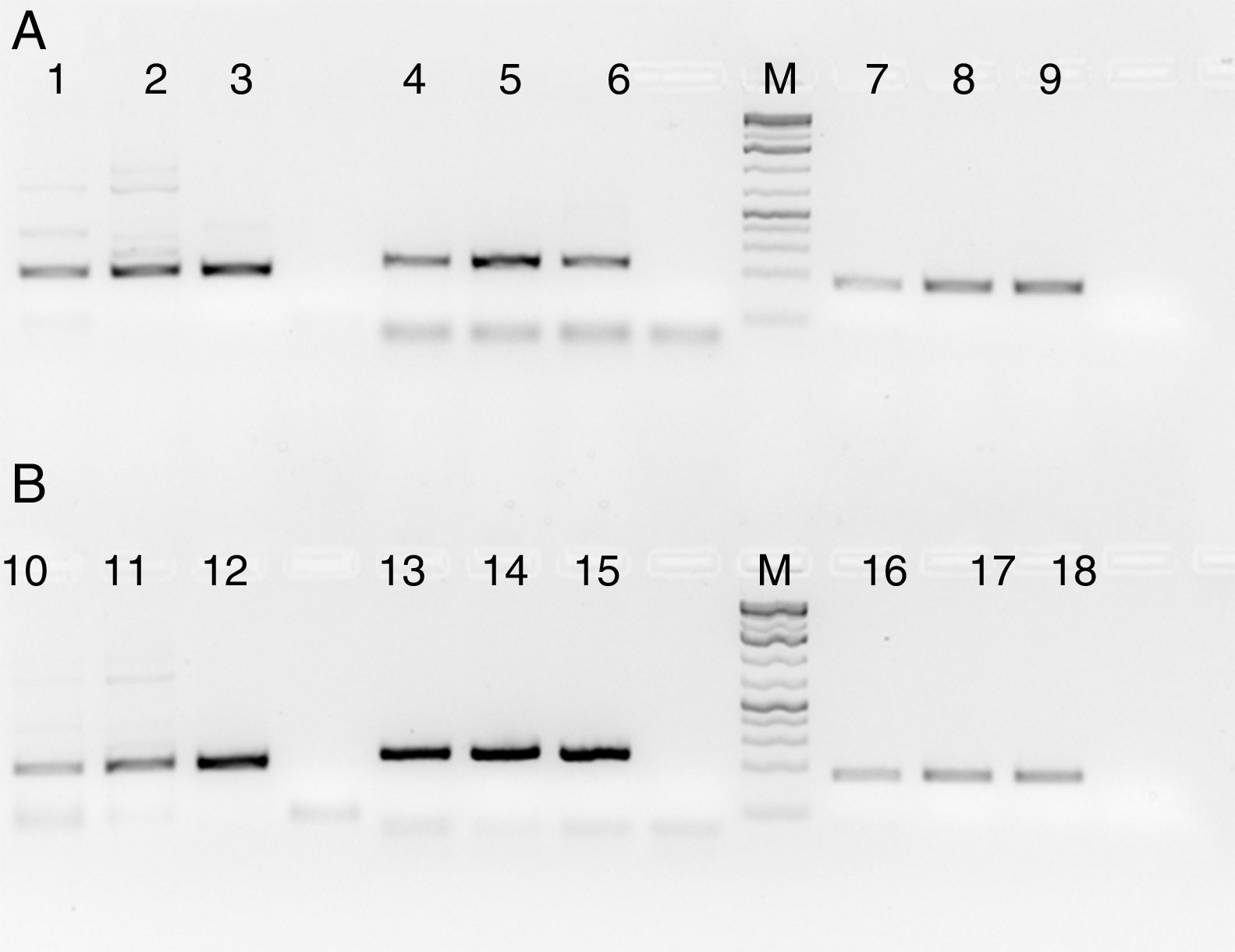
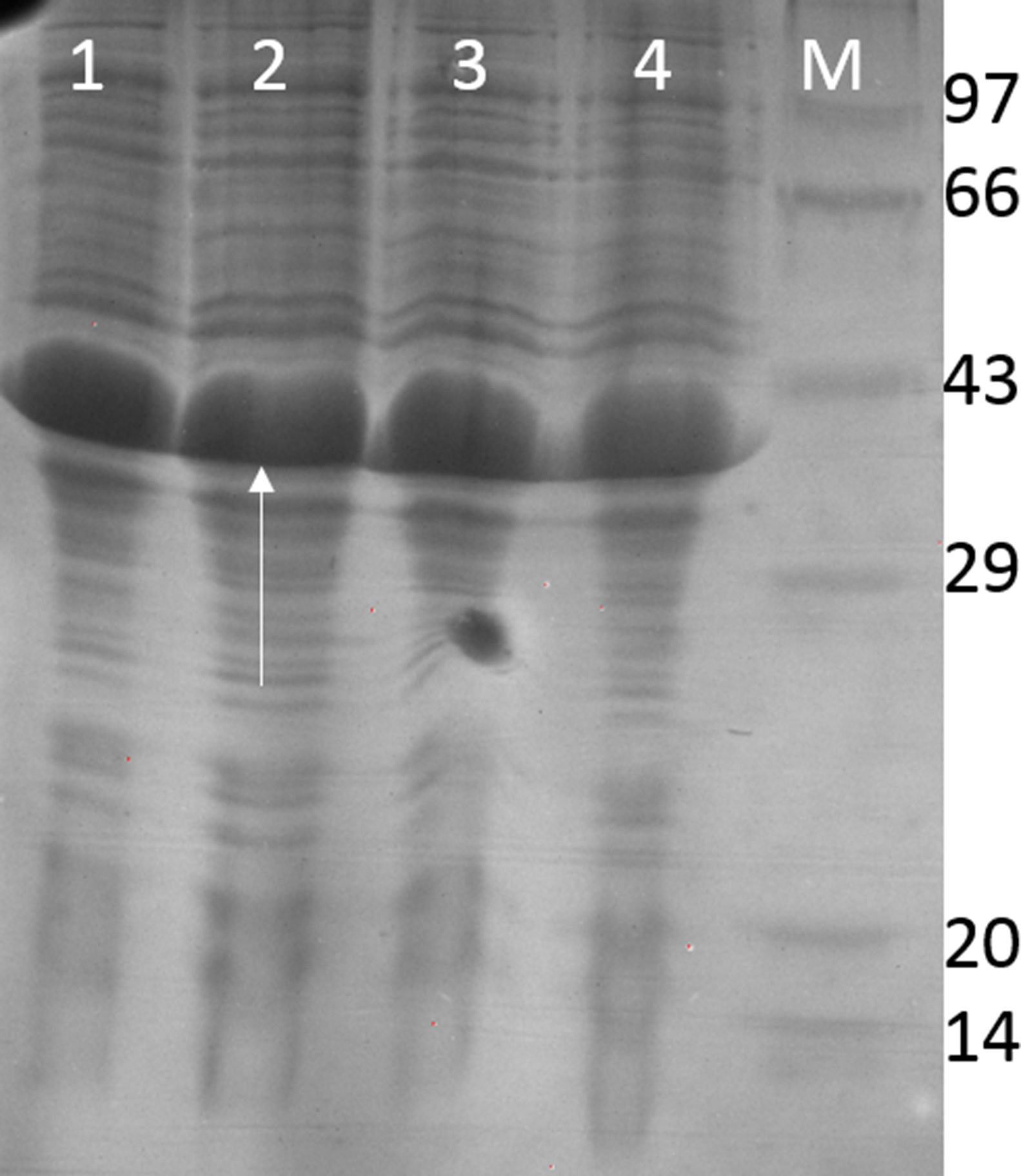
Fig. 4 shows the PCR amplification of HIV/HBV and HCV target genes using three ELISA positive clinical samples. It is evident from Fig. 4 that RecA improved the PCR signal intensities of the HBV samples (Fig. 4, lanes 13, 14 and 15 in comparison to lanes 4, 5 and 6), while the PCR signals of target genes of RNA viruses such as HIV and HCV did not show any improvements, albeit there was marked reduction in levels of non-specific amplifications when RecA was included in the PCR reaction mix. Since HBV is a DNA virus, it is possible to speculate that RecA enhances the annealing of the HBV specific primers to the target S gene of the HBV, and due to the reported role of RecA in minimizing false priming, the intensity of the PCR signals for the HBV samples appear higher with RecA in comparison to the PCR of the same samples without RecA.

PCR products of three HIV, HBV and HCV samples with and without RecA. Panel A shows PCR without RecA, while panel B shows PCR with RecA. Lanes 1, 2, 3, 10, 11 and 12 are HIV samples; lanes 4, 5, 6, 13, 14, and 15 are HBV samples; lanes 7, 8, 9, 16, 17, and 18 are HCV samples. RT-PCR was performed for the HIV and HCV samples as described in the M & M section, while for HBV, regular PCR was performed as per the protocol described in the M & M section.
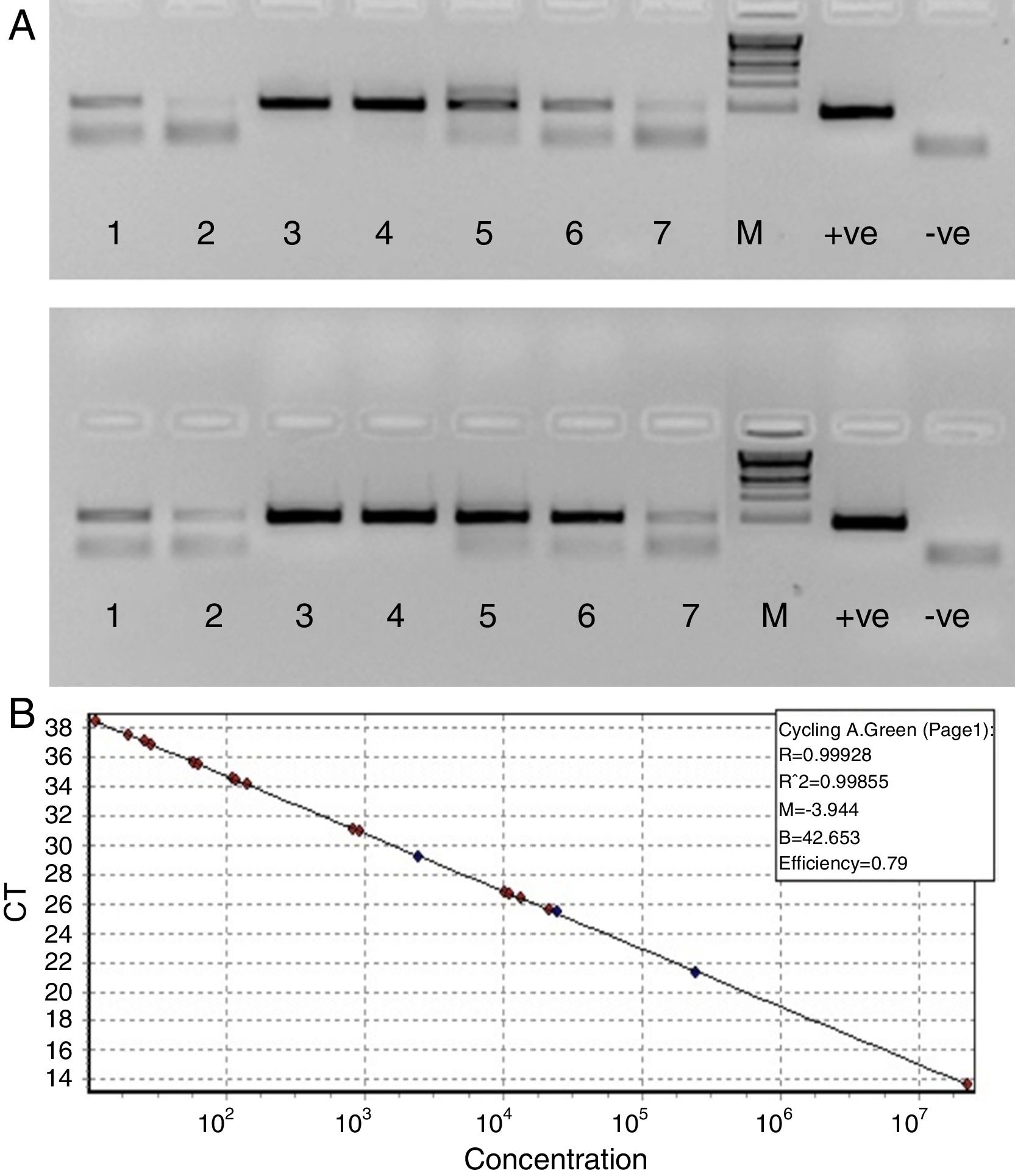
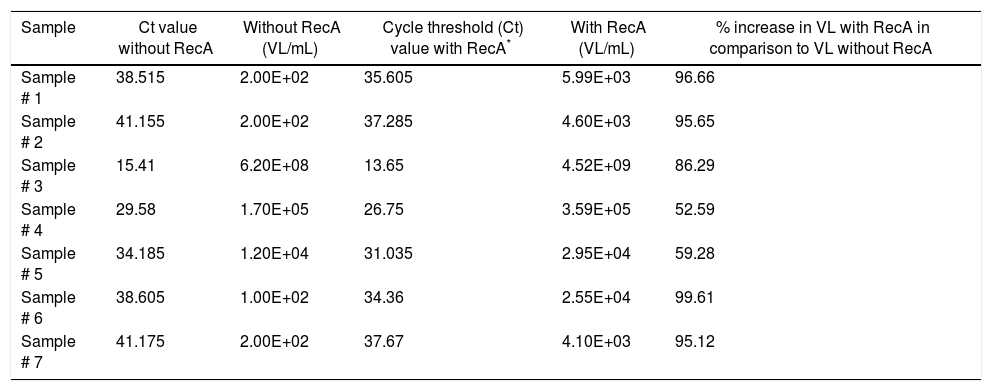
Since our data showed that the PCR signals of HBV was higher in presence of RecA in comparison to that achieved without RecA, we wanted to examine if RecA would also have an effect on the HBV viral load values in the HBV positive plasma samples. From Fig. 5A, it is clear that the sensitivity of the HBV detection is enhanced and the overall HBV viral load values were higher (by 50–98%) for all the samples tested in this study when RecA was included in the PCR reaction mixture in comparison to the HBV viral load values obtained for the same samples without RecA (Table 1). Fig. 5B shows the standard curve prepared using plasmid pTZ57R/T-HBV from 250 to 2.5×105copies/mL.
 Agarose gel depicting PCR of HBV samples. Panel A shows PCR without RecA, while panel B shows PCR with RecA for the same samples. Lanes 1, 2, 3, 4, 5, 6 and 7 show PCR amplicons from ELISA positive HBV samples. +ve denotes positive control PCR with pTZ57R/T-HBV plasmid and −ve denotes PCR with no template control. (B) Representative standard curve of the SYBR Green-1 qPCR assay for HBV. The plots were generated using a 10-fold dilution series ranging from 250 to 2.5×105copies/mL of plasmid DNA standard (pTZ57R/T). Duplicate reactions for each standard dilution were used. The standard curve was generated by plotting the log10 copy number of starting plasmid quantity on the x-axis against the corresponding threshold cycle (Ct) value on the y-axis.")
(A) Agarose gel depicting PCR of HBV samples. Panel A shows PCR without RecA, while panel B shows PCR with RecA for the same samples. Lanes 1, 2, 3, 4, 5, 6 and 7 show PCR amplicons from ELISA positive HBV samples. +ve denotes positive control PCR with pTZ57R/T-HBV plasmid and −ve denotes PCR with no template control. (B) Representative standard curve of the SYBR Green-1 qPCR assay for HBV. The plots were generated using a 10-fold dilution series ranging from 250 to 2.5×105copies/mL of plasmid DNA standard (pTZ57R/T). Duplicate reactions for each standard dilution were used. The standard curve was generated by plotting the log10 copy number of starting plasmid quantity on the x-axis against the corresponding threshold cycle (Ct) value on the y-axis.
Hepatitis B virus (HBV) viral load values of enzyme-linked immune sorbent assay (ELISA) positive HBV samples with and without RecA.
| Sample | Ct value without RecA | Without RecA (VL/mL) | Cycle threshold (Ct) value with RecA* | With RecA (VL/mL) | % increase in VL with RecA in comparison to VL without RecA |
|---|---|---|---|---|---|
| Sample # 1 | 38.515 | 2.00E+02 | 35.605 | 5.99E+03 | 96.66 |
| Sample # 2 | 41.155 | 2.00E+02 | 37.285 | 4.60E+03 | 95.65 |
| Sample # 3 | 15.41 | 6.20E+08 | 13.65 | 4.52E+09 | 86.29 |
| Sample # 4 | 29.58 | 1.70E+05 | 26.75 | 3.59E+05 | 52.59 |
| Sample # 5 | 34.185 | 1.20E+04 | 31.035 | 2.95E+04 | 59.28 |
| Sample # 6 | 38.605 | 1.00E+02 | 34.36 | 2.55E+04 | 99.61 |
| Sample # 7 | 41.175 | 2.00E+02 | 37.67 | 4.10E+03 | 95.12 |
The T7 promoter is known to be the strongest promoter known for the prokaryotic expression of proteins35 and in experimental E. coli cultures, consistently higher expression levels of the protein have been observed with the kanamycin marker than in cultures grown with the commonly used antibiotic ampicillin36 with the target protein being almost 50% of total cellular protein in some cases.37 It is possible to speculate that for the above reasons, we achieved high levels of RecA expression described in this study.
One could observe a small fraction of RecA protein induced even under uninduced conditions (Fig. S4, lane 1). Expression of RecA under uninduced conditions could be attributed to the leaky expression of the T7 promoter that is seen commonly in E. coli cells and can be easily controlled using plysE plasmid, which expresses T7 lysozyme, an inhibitor of T7 RNA polymerase.38 Also, one could see appreciable amounts of RecA expressed even with the lowest concentration of IPTG (0.125mM) which supports the recent report of high level expression of a leptospiral protein in E. coli using such low concentrations of IPTG.39
A number of protocols have been reported for purification of wild-type RecA protein from E. coli or Thermus aquaticus. These methods include salt precipitation followed by hydrophobic interaction chromatography and anion exchange column,25 precipitation using spermidine,40 purification using chitin affinity column,41 monoclonal antibody affinity columns,42 and phosphocellulose matrix.43 Many of these protocols require multiple steps for purification and are not very cost-effective. In an effort to develop a simple and a cost-effective protocol for RecA purification, we took advantage of the heat stable property of RecA and obtained highly pure RecA preparations by a simple heat treatment of the crude lysate without the use of any chromatographic steps and such a methodology for the purification of RecA is hitherto unreported.
A significant amount of endonuclease contamination from the host endA gene has been reported in RecA protein preparations by Singleton et al.,41 which was removed by rigorous washing of the induced cells with detergents. The protocol described in this article utilizes an endA1 negative E. coli host, ER2566, for RecA expression, and hence, our purified RecA is virtually free of any endonuclease contamination, which is a huge advantage for its use in PCR-based diagnostics.
The RecA gene of T. thermophilus encodes a 36kDa polypeptide whose amino acid sequence shows 61% identity with that of the E. coli RecA protein and T. thermophilus RecA is reported to be thermostable in comparison to E. coli RecA protein27 with similar functions. The yield of recombinant RecA (from E. coli or T. thermophilus) from various literature reports is variable and not feasible for commercial scale operations. While the protocol described by Griffith and Shores40 yield only 10mg of RecA protein per liter, the protocol developed by Singleton et al.41 showed a yield of 28mg of RecA/liter of E. coli culture. In this study, we have achieved 35mg of pure RecA per liter of E. coli culture, which is the highest report of RecA yield till date. Additionally, the cost of 1mg of commercial RecA is $575 and we are able to produce almost US$20,125 worth RecA from every liter of E. coli culture, which is interesting and useful.
Lactose can also be used as an inducer during the transition from the exponential to the stationary phase for protein induction for genes cloned under T7 promoter. Since lactose also acts as carbon source, the use of lactose for induction would prove useful for increasing the RecA expression per cell.44
ATP hydrolysis serves as a means of releasing RecA promoters from the 5′ end of the filament once DNA strand exchange occurs, thus helps recycle the RecA protein.45 Additionally, it has been shown by Yu and Egelman46 that the RecA filament is inactive when the compressed filament is formed in the absence of a nucleotide cofactor such as ATP, either as a self-polymer or on a single-stranded DNA molecule. Hence, we hypothesized that ATP hydrolysis is a crucial step for the activity of RecA, and hence, the ATPase activity of RecA can serve as a measure for monitoring RecA strand exchange activity. We observed that the ATPase activity of the in-house RecA and commercial RecA preparations were substantially similar. This data indicates that the purified RecA does not have the property of aggregation, since the aggregation state of RecA protein is known to affect its ATPase activity.47
Shigemori et al.24 have performed RT-PCR for their target gene using a SYBR premix Ex Taq kit with and without RecA and observed a nearly 30% increase in PCR fluorescence signal intensity with RecA compared to that without RecA. Our observations of ∼50 to 96% increase in the fluorescence intensity of the HBV PCRs with RecA supports the reported observations. In addition, we show lower Ct values in HBV qPCR with RecA, which is a new observation. Also, the limit of detection (LOD) of the HBV viral load method developed using SYBR green dye appears to be 5viruscopies/reaction which equates to 40IU/mL. This LOD value is very close to the LOD value of the Abbott HBV viral load kit (utilizing TaqMan chemistry) of 15IU/mL for a procedure processing 0.2mL of the starting sample.
An assay with a lower limit of detection of 103IU/mL of HBV is reported to be sufficient to monitor and manage the patient,48 but in some instances, a more sensitive assay with a lower limit of detection on the order of 101IU/mL is recommended to ensure detection of the emergence of resistance as early as possible. Hence, our observations of enhancement of the limit of HBV viral load detection by almost 1log copies can be extrapolated to improving the qPCR HBV LOD from 200copies/mL to 20copies/mL or 40IU/mL to 4IU/mL, which is hitherto unreported.
In this paper, we have described a real-time PCR assay for HBV DNA quantification with SYBR Green chemistry. The primers used for qPCR targets the surface antigen of HBV and have been designed after aligning nucleotide sequences of all the HBV genotypes available in the NCBI database so that one is assured of positive PCR signal for all HBV serotypes using these set of primers.
Viral load set point has long been used as a prognostic marker of disease progression and monitoring of virus load is known to predict the possible emergence of drug-resistant HBV strains. Also, an undetectable or low viral load does not necessarily guarantee that a patient will not experience liver damage; hence methods that enhance the sensitivity of the viral load method would have immense value in clinical setting and further drug treatments. In the light of this, our observation of RecA enhancing the LOD of the viral load estimation method is novel and interesting.
We are presently focusing on generating novel RecA fusions to examine whether the heat stability of RecA can be imparted to heterologous genes and whether such heterologous RecA fusion proteins could be purified by heat treatment as described in this article.
Conflicts of interestThe authors declare no conflicts of interest.
The authors wish to thank Dr. C. Nagaraj, Prof. of Microbiology, PESIMSR, Kuppam, India, for the supply of clinical samples of HIV, HBV and HCV for our studies. The authors also wish to thank Dr. Sridhar, K.N. and Dr. Gautham Nadig for their constant support and encouragement. Cancyte Technologies Pvt. Ltd. is fully supported by Sri Sringeri Sharada Peetam, India.