



La craneosinostosis es una anomalía congénita resultante de la fusión prematura de las suturas craneales que cambia los patrones de crecimiento del cráneo.
MetodologíaSe definió el enfoque, los alcances, la población blanco y las preguntas clínicas a resolver. Se realizó una búsqueda sistematizada de la evidencia por etapas; en la primera se buscaron guías de práctica clínica, en la segunda, revisiones sistemáticas, y en la tercera, ensayos clínicos y estudios observacionales en Medline, Embase, KoreaMed, Cochrane Library y el portal de la Organización Mundial de la Salud utilizando los términos MeSH, Decs y libres correspondientes, sin restricciones de lenguaje ni temporalidad. Se evaluó el riesgo de sesgo de cada documento utilizando las herramientas AMSTAR, Risk of Bias y STROBE. Se graduó la calidad de la evidencia utilizando el sistema GRADE. Se utilizó la técnica de Panel Delphi modificada para asignar la dirección y la fuerza de la recomendación, así como el grado de acuerdo con la misma, tomando en cuenta para esto la calidad de la evidencia, el balance entre riesgos y beneficios de las intervenciones, los valores y las preferencias de los pacientes y la disponibilidad de los recursos.
ResultadosSe obtuvieron mediante los algoritmos de búsqueda 3,712 documentos, seleccionando para la inclusión en la síntesis cualitativa 29. Debido a la heterogeneidad entre los estudios no fue posible realizar un metaanálisis.
ConclusionesSe emitieron 7 recomendaciones y 8 puntos de buena práctica, los cuales servirán como apoyo para la toma de decisiones en la atención integral de pacientes con craneosinostosis.
Craniosynostosis is a congenital anomaly resulting from the premature fusion of the cranial sutures changing growth patterns of the skull.
MethodologyFocus, scope, target population and clinical questions to be solved were defined. A systematic search for evidence in different databases (Medline, Embase, KoreaMed, Cochrane Library and the website of the World Health Organization) in stages was performed: clinical practice guidelines; systematic reviews, and clinical trials and observational studies; using MeSH, Decs and corresponding free terms, unrestricted language or temporality. Risk of bias was evaluated using appropriate tools (AMSTAR, Risk of Bias or STROBE). The quality of evidence was graduated using the GRADE system. Modified Delphi Panel technique was used to assign the recommendation's strength and direction, as well as the degree of agreement with it, taking into account the quality of evidence, balance between risks and benefits of interventions, values and preferences of patients and availability of resources.
ResultsThere were 3,712 documents obtained by search algorithms; selecting 29 documents for inclusion in the qualitative synthesis. Due to heterogeneity between studies, it was not possible to perform meta-analysis.
ConclusionsWe issued 7 recommendations and 8 good practice points, which will serve as support for decision-making in the comprehensive care of patients with craniosynostosis.
La craneosinostosis es una anomalía congénita común, resultante de la fusión prematura de las suturas craneales, que cambia los patrones de crecimiento del cráneo1. Se clasifica en simple o compuesta (dependiendo de si afecta una o varias suturas) y en primarias y secundarias. Las primarias son de causa genética y con frecuencia están presentes desde el nacimiento. También se dividen en sindrómicas (familiares, hereditarias) y no sindrómicas (aisladas). Las secundarias son por un trastorno adquirido causado por una enfermedad conocida, como microcefalia, talasemia, anemia de células falciformes, trastornos metabólicos o teratógenos, entre otros. Los casos no sindrómicos son los más frecuentes; algunos pueden ser de origen genético, pero no tienen una herencia mendeliana. Las craneosinostosis sindrómicas de causa genética representan del 10 al 20% de los casos. Se han descrito más de 100 síndromes asociados a craneosinostosis. La incidencia global se ha calculado en uno por 2,000 a 2,500 recién nacidos vivos; la prevalencia de todos los tipos de craneosinostosis, aisladas y sindrómicas, es de 343 por un millón. La incidencia de craneosinostosis no sindrómica es de aproximadamente 0.6 por 1,000 nacidos vivos2.
La sutura sagital es afectada en un 40 a un 60%, la sutura coronal en un 20 a un 30% y la sutura metópica en menos del 10% de los casos; la sinostosis lambdoidea verdadera es rara3.
Comúnmente, la craneosinostosis está presente al nacimiento, pero no siempre se diagnostica cuando es leve. Usualmente es diagnosticada como una deformidad craneal durante los primeros meses de vida. El diagnóstico se basa en la exploración física (observando la forma del cráneo y la cara, además de palpando los bordes de las suturas y las fontanelas) y en estudios radiológicos, incluyendo radiografía y tomografía computada de cráneo2,4.
Puede ser difícil de reconocer el cierre prematuro de las suturas, por lo que es preciso vigilar el perímetro cefálico para llegar a identificar oportunamente la craneosinostosis5.
La craneosinostosis puede derivar en 2 grupos de problemas principales. Puede observarse un incremento en la presión intracraneana con o sin hidrocefalia, aunque esto es más común cuando están involucradas múltiples suturas. También puede encontrarse deformación ósea tanto de cráneo como facial. Para la corrección estética o la disminución de la presión intracraneana, la cirugía correctiva temprana está indicada porque el 50% del crecimiento del cráneo se consigue a los 6 meses de edad3.
Es importante diferenciar la sinostosis lambdoide de la plagiocefalia (también llamada plagiocefalia occipital, plagiocefalia posterior y plagiocefalia sin sinostosis), que es debida a una deformación postural. La incidencia de la plagiocefalia es de aproximadamente uno por cada 300 nacidos vivos, comparado con la rara incidencia de la sinostosis lambdoidea6.
La evaluación temprana de un niño con craneosinostosis es imperativa y puede iniciarse perinatalmente. Un equipo interdisciplinario es importante porque el momento adecuado de la intervención es crítico. Por otra parte, la atención coordinada es necesaria por la complejidad de los factores médicos, quirúrgicos y psicosociales. Mientras el manejo temprano puede derivar en mejores resultados (por ejemplo, pocas operaciones y bajos costos), la continuidad del cuidado en equipo es esencial porque los resultados son medidos a través del crecimiento y el desarrollo del niño4.
En este momento, a nivel nacional o internacional no existe una guía de práctica clínica para el diagnóstico, tratamiento o rehabilitación de pacientes con craneosinostosis, por lo que el objetivo de este trabajo es realizar la primera guía referente a este tema. Esta guía forma parte del Catálogo Maestro de Guías de Práctica Clínica, cuya finalidad es establecer un referente nacional para orientar la toma de decisiones clínicas basadas en recomendaciones sustentadas en la mejor evidencia disponible. Lo anterior favorecerá la mejora en la efectividad, la seguridad y la calidad de la atención médica, contribuyendo, de esta manera, al bienestar de las personas y de las comunidades, lo que constituye el objetivo central y la razón de ser de los servicios de salud. El Catálogo Maestro de Guías de Práctica Clínica se aloja en el sitio web del Centro Nacional de Excelencia Tecnológica en Salud (http://www.cenetec.salud.gob.mx/contenidos/gpc/catalogoMaestroGPC.html#).
Los impactos esperados derivados de la realización y la implementación de esta guía son: aumento de la tasa de diagnóstico temprano, aplicación de tratamiento oportuno, disminución de la tasa de secuelas y mejoría en la calidad de vida de los pacientes con craneosinostosis.
Recomendaciones- •
Se recomienda realizar una exploración física dirigida para descartar craneosinostosis no sindrómica a los recién nacidos con factores de riesgo. Baja calidad de evidencia/Recomendación fuerte.
- •
No existe suficiente evidencia para recomendar realizar diagnóstico prenatal de craneosinostosis por ultrasonido. Muy baja calidad de evidencia/Sin recomendación.
- •
Se recomienda realizar radiografía de cráneo (anteroposterior y lateral) y tomografía craneal con reconstrucción 3D en pacientes con sospecha de craneosinostosis. Baja calidad de evidencia/Recomendación fuerte.
- •
Se recomienda que a los niños con sinostosis bicoronal o unicoronal confirmada se les realice un estudio genético molecular que incluya las mutaciones en FGFR2 exones iiia/iiic, FGFR3 (P250R) y TWIST1 para determinar la etiología. Baja calidad de evidencia/Recomendación fuerte.
- •
Se recomienda realizar tratamiento quirúrgico antes de los 12 meses de edad en pacientes con craneosinostosis. Muy baja calidad de evidencia/Recomendación fuerte.
- •
Se recomienda realizar el tratamiento quirúrgico de acuerdo con el tipo de craneosinostosis. Muy baja calidad de evidencia/Recomendación fuerte.
- •
Se aconseja realizar suturectomía y remodelación craneal o distractores tipo Spring en pacientes con escafocefalia. Muy baja calidad de evidencia/Recomendación débil.
- 1.
Se recomienda considerar la búsqueda intencionada de craneosinostosis no sindrómica a través de ultrasonido prenatal cuando existan factores de riesgo.
- 2.
Se recomienda determinar el índice craneal de forma pre y postoperatoria.
- 3.
Se recomienda brindar asesoramiento genético a los padres de pacientes con craneosinostosis no sindrómica con la finalidad de que comprendan los aspectos médicos de la enfermedad, la etiología, el curso probable de la misma, el manejo posible, así como el riesgo de recurrencia para una correcta toma de decisiones.
- 4.
Se recomienda realizar la técnica de avance frontoorbitario para pacientes con trigonocefalia y plagiocefalia anterior.
- 5.
Todo paciente con sospecha de craneosinostosis no sindrómica debe referirse a Neurocirugía o Cirugía Plástica para confirmación diagnóstica y tratamiento.
- 6.
Todo paciente con diagnóstico de craneosinostosis no sindrómica se debe enviar a Genética para determinar la etiología y brindar asesoramiento genético.
- 7.
Todo paciente con diagnóstico de craneosinostosis no sindrómica se debe enviar a Rehabilitación para vigilancia del neurodesarrollo e intervención temprana para evitar secuelas neurológicas.
El grupo desarrollador de la guía de práctica clínica para el diagnóstico, tratamiento y rehabilitación de craneosinostosis no sindrómica en los 3 niveles de atención se conformó por profesionales de la salud especialistas en medicina física y rehabilitación, genética, neurocirugía, cirugía general y medicina interna, así como licenciados en terapia física y rehabilitación, quienes de manera conjunta definieron el enfoque, los alcances y las preguntas clínicas de la guía. La población blanco fueron niños de 0 a 23 meses de edad.
Las preguntas clínicas consideradas para esta guía son:
- 1.
¿Cuáles son los factores de riesgo para presentar craneosinostosis no sindrómica?
- 2.
¿Cuáles son los criterios de referencia para los pacientes con craneosinostosis no sindrómica?
- 3.
¿Es útil el diagnóstico prenatal de craneosinostosis no sindrómica?
- 4.
¿Cuál es el estudio de gabinete más útil para el diagnóstico de craneosinostosis no sindrómica?
- 5.
¿Cuál es la utilidad de los estudios genéticos en craneosinostosis no sindrómica?
- 6.
¿Es útil el asesoramiento genético para los pacientes con craneosinostosis no sindrómica?
- 7.
¿Cuál es la utilidad del tratamiento quirúrgico temprano de los pacientes con craneosinostosis no sindrómica?
- 8.
¿Cuál es el tratamiento quirúrgico de craneosinostosis no sindrómica?
- 9.
¿Cuál es la utilidad del manejo de rehabilitación en pacientes con craneosinostosis no sindrómica para evitar secuelas neurológicas?
Se realizó una búsqueda sistematizada de la evidencia. En la primera etapa se buscaron guías de práctica clínica en Medline, así como en los siguientes sitios electrónicos: National Institute for Health and Care Excellence, Trip Database, Asociación Médica Canadiense, Scottish Intercollegiate Guidelines Network, Ministerio de Salud de Chile, Biblioteca del Sistema Nacional de Salud de España y National Guideline Clearinghouse.
En la segunda etapa se buscaron revisiones sistemáticas en las bases de datos: Medline, Embase, así como en Cochrane Library. En la tercera etapa se realizó una búsqueda de estudios primarios específica para cada pregunta utilizando los términos MeSH y Decs correspondientes a cada pregunta en formato PICO, sin restricciones de lenguaje ni temporalidad de la guía en las bases de datos: Medline, Embase, KoreaMed, así como la Cochrane Library y el portal de la Organización Mundial de la Salud.
Se revisaron todos los artículos obtenidos a través de los algoritmos de búsqueda de las diferentes bases de datos por título y resumen. Se seleccionaron aquellos documentos referentes al tema y se analizaron a texto completo seleccionando aquellos que cumplían los criterios de inclusión para realizar la síntesis de la información.
Se evaluó el riesgo de sesgo de cada documento utilizando la herramienta AMSTAR7 para las revisiones sistemáticas, «Risk of Bias8 de Cochrane para el ensayo clínico y STROBE9 para los estudios observacionales. Se graduó la calidad de la evidencia utilizando el sistema GRADE10, declarando junto con la evidencia la calidad asignada.
Se elaboró un borrador de las recomendaciones por el grupo desarrollador de la guía y posteriormente se realizó una técnica de Panel Delphi modificada11 para asignar la dirección y fuerza de la recomendación, así como el grado de acuerdo con la misma, tomando en cuenta para esto la calidad de la evidencia, el balance entre riesgos y beneficios de las intervenciones, los valores y preferencias de los pacientes y la disponibilidad de los recursos, obteniendo una aprobación mayor del 80% en todas ellas. En el caso de las preguntas en la que no se encontró evidencia suficiente para realizar una recomendación se elaboró un punto de buena práctica clínica, el cual también se consensó con un grado de acuerdo del 100%.
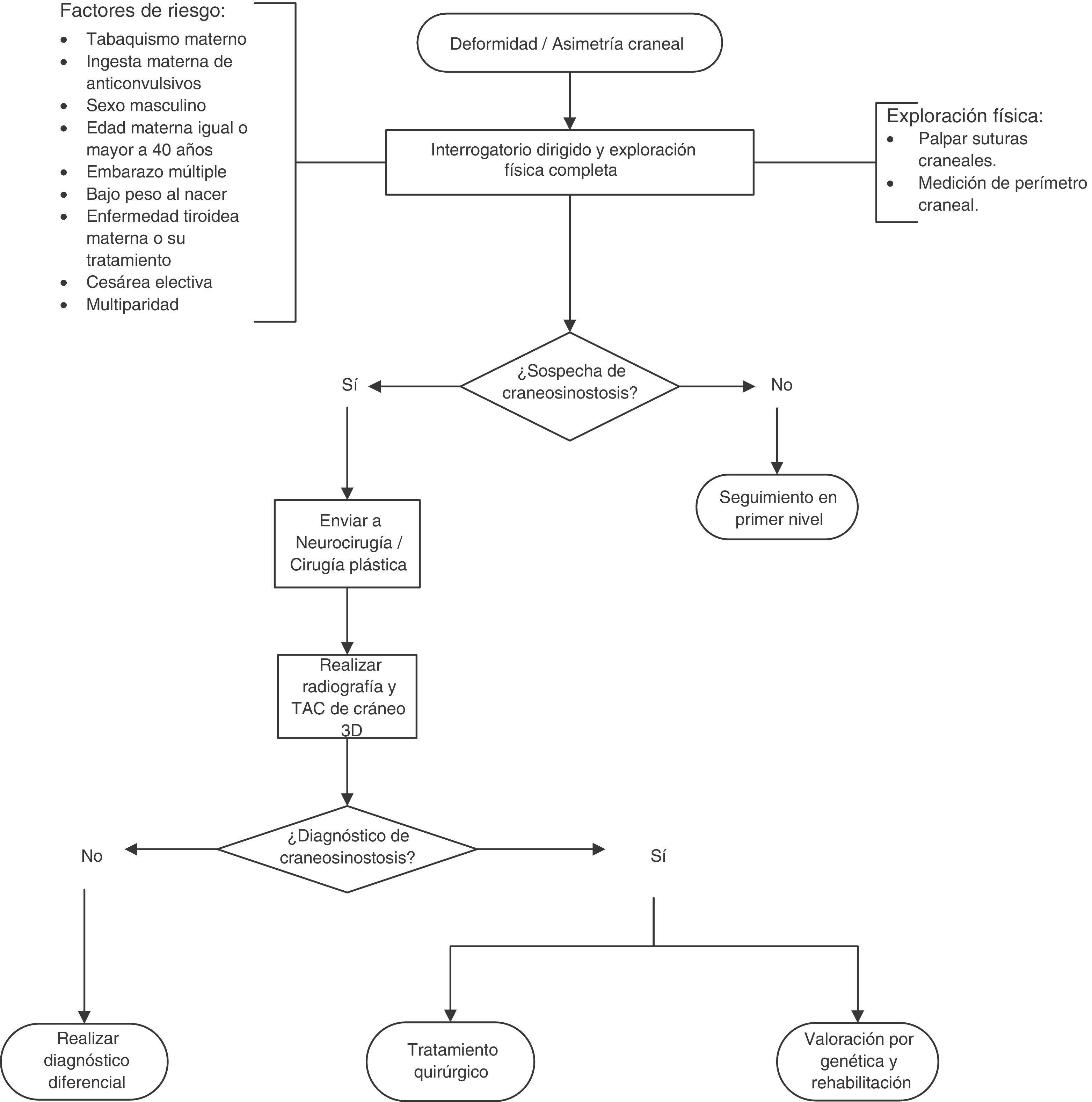
Se realizó un diagrama de flujo del diagnóstico y el tratamiento de la craneosinostosis en niños de 23 meses o menos, basado en las recomendaciones y los puntos de buena práctica (fig. 1).

Se eligieron por parte del grupo elaborador las recomendaciones más importantes de acuerdo con su impacto esperado en los objetivos de la guía.
Una vez terminado el borrador final de la guía se realizó la validación de contenido por un par clínico experto en el tema, la verificación interinstitucional por el Instituto Mexicano del Seguro Social y la posterior autorización por el Comité Nacional de Guías de Práctica Clínica.
Esta guía será actualizada cuando exista evidencia que así lo determine o de manera programada a los 3-5 años de la publicación. Se estableció una monitorización continua de la guía por parte de los usuarios mediante el blog: http://cenetec-difusion.com/gpc-sns/
Esta guía pone a disposición del personal del primer, segundo o tercer niveles de atención las recomendaciones basadas en la mejor evidencia disponible con la intención de estandarizar las acciones acerca de:
- •
Identificar los factores de riesgo para presentar craneosinostosis no sindrómica.
- •
Especificar los criterios de referencia para los pacientes con craneosinostosis no sindrómica.
- •
Valorar la utilidad del diagnóstico prenatal para craneosinostosis no sindrómica.
- •
Determinar el estudio de gabinete más útil para el diagnóstico de craneosinostosis no sindrómica.
- •
Evaluar la utilidad de los estudios genéticos en craneosinostosis no sindrómica.
- •
Determinar la conveniencia del asesoramiento genético para los pacientes con craneosinostosis no sindrómica.
- •
Identificar el tratamiento quirúrgico de craneosinostosis no sindrómica.
- •
Establecer la utilidad del tratamiento quirúrgico temprano de los pacientes con craneosinostosis no sindrómica.
- •
Evaluar la utilidad del manejo de la rehabilitación en pacientes con craneosinostosis no sindrómica para evitar secuelas neurológicas.
Los usuarios diana de esta guía son estudiantes de Medicina, enfermeras generales, médicos pasantes en servicio social, médicos generales, médicos residentes, médicos familiares, médicos especialistas en pediatría, genética y neurocirugía.
ResultadosSe obtuvieron mediante los algoritmos de búsqueda 3,712 documentos, los cuales se comprobaron por título y resumen revisando a texto completo aquellos referentes a la pregunta PICO «específica», seleccionando para la inclusión en la síntesis cualitativa 29 documentos. De estos, 2 fueron revisiones sistemáticas, uno, un ensayo clínico aleatorizado y 26 fueron estudios observacionales. También se utilizaron 5 documentos metodológicos y para fundamentar la definición y la justificación. Debido a la heterogeneidad entre los estudios no fue posible realizar un metaanálisis.
Recomendaciones y resumen de evidenciasRecomendación: se recomienda realizar una exploración física dirigida para descartar craneosinostosis no sindrómica a los recién nacidos con factores de riesgo. Baja calidad de evidencia/Recomendación fuerte.
Evidencia: los factores de riesgo en los que se ha encontrado asociación estadísticamente significativa para presentar craneosinostosis son (tabla 1)12–19:
Factores de riesgo asociados a craneosinostosis
| Factor de riesgo | Lee et al.17 | Barik et al.13 | Alderman et al.12 | Källén16 | Zeiger et al.19 | Carmichael et al.14 | Källén y Robert-Gnansia15 | Rasmussen et al.18 |
|---|---|---|---|---|---|---|---|---|
| Sexo masculino | OR 2.0 (IC 1.64-2.44) | – | OR 1.6 (IC 1.1-2.2) | OR 2.8 (IC 1.7-2.8) | – | – | – | – |
| Embarazo múltiple | OR 2.36 (IC 1.62-3.44) | – | OR 3.0 (IC 1.2-7.1) | – | – | – | – | – |
| Tabaquismo materno durante el embarazo | – | – | – | OR 1.45 (IC 1.13-1.87) | Sin diferencia estadística | OR 8.0 (IC 2.2-25.6) | – | – |
| Parto prematuro | OR 2.21 (IC 1.68-2.91) | OR 1.7 (IC 1.2-2.5) y OR 2.8 (IC 1.6-4.7) | – | – | – | – | – | – |
| Bajo peso al nacer (1,500-2,499g) | OR 1.97 (IC 1.41-2.75) | – | – | – | – | – | – | – |
| Muy bajo peso al nacer (menos de 1,500g) | OR 4.37 (IC 2.64-7.21) | – | – | – | – | – | – | – |
| Bajo peso para la edad gestacional | OR 1.64 (IC 1.25-2.14) | – | – | – | – | – | – | – |
| Edad materna mayor o igual a 40 años | OR 1.92 (IC 1.17-3.15) | – | – | – | – | – | – | – |
| Cesárea electiva | OR 1.71 (IC 1.31-2.23) | – | – | – | – | – | – | – |
| Multiparidad | – | – | – | OR 2.0 (IC 1.2-2.4) | – | – | – | – |
| Enfermedad tiroidea materna o su tratamiento | – | – | – | – | – | – | – | OR 2.47 (IC 1.46-4.18) |
| Uso materno de anticonvulsivos | – | – | – | – | – | – | RR 6.9 (IC 1.10-7.94) | – |
| Nuliparidad | – | OR 2.0 (IC 1.1-2.7) | – | – | – | – | – | – |
| Macrosomía | – | OR 2.0 (IC 1.2-2.4) | – | – | – | – | – | – |
Del producto:
- •
Sexo masculino.
- •
Muy bajo peso al nacer (<1,500g).
- •
Bajo peso al nacer (1,500 a 2,499g).
- •
Bajo peso para la edad gestacional.
- •
Macrosomía.
Maternos:
- •
Edad≥40 años.
- •
Tabaquismo durante el embarazo.
- •
Enfermedad tiroidea o su tratamiento.
- •
Uso de anticonvulsivos.
- •
Embarazo múltiple.
- •
Multiparidad.
- •
Nuliparidad.
- •
Cesárea electiva.
- •
Parto prematuro.
Recomendación: no existe suficiente evidencia para recomendar realizar diagnóstico prenatal de craneosinostosis por ultrasonido20–22. Muy baja calidad de evidencia/Sin recomendación.
Punto de buena práctica: se aconseja considerar la búsqueda intencionada de craneosinostosis no sindrómica a través de ultrasonido prenatal cuando existan factores de riesgo.
Evidencia: en un estudio de serie de casos se evaluó la utilidad del ultrasonido prenatal para el diagnóstico temprano de craneosinostosis en fetos con factores de riesgo, determinado por la disminución del espacio de las suturas craneales, encontrando una sensibilidad del 100% y una especificidad del 97% para el ultrasonido20. Otro estudio de serie de casos reportó una sensibilidad del 58%, aunque el diagnóstico de craneosinostosis se basó en la geometría craneal21. En un estudio de serie de casos de 15 fetos con ultrasonido prenatal anormal se realizó resonancia magnética, encontrando un 100% de sensibilidad y de especificidad al correlacionarse con un seguimiento y diagnóstico posnatal22.
Recomendación: se recomienda realizar radiografía de cráneo (anteroposterior y lateral) y tomografía craneal con reconstrucción 3D en pacientes con sospecha de craneosinostosis. Baja calidad de evidencia/Recomendación fuerte.
Punto de buena práctica: se aconseja determinar el índice craneal de forma pre y postoperatoria23.
Evidencia: la sensibilidad de las radiografías llega a ser del 80% con una especificidad del 95%, mientras que la sensibilidad de la tomografía con reconstrucción en 3D es del 96% y la especificidad del 100%. La centellografía ósea esta fuera de uso debido a su baja precisión global diagnóstica, estimada en un 66%. En un estudio de prueba diagnóstica se comparó la precisión diagnóstica del ultrasonido frente a la tomografía en 44 pacientes, encontrando que el ultrasonido tiene una sensibilidad del 96.9%, una especificidad del 100%, un valor predictivo positivo del 100% y un valor predictivo negativo del 92.3%24–27.
Recomendación: se recomienda que a los niños con sinostosis bicoronal o unicoronal confirmada se les realice un estudio genético molecular que incluya las mutaciones en FGFR2 exones iiia/iiic, FGFR3 (P250R) y TWIST1 para determinar la etiología. Baja calidad de evidencia/Recomendación fuerte.
Punto de buena práctica: se aconseja brindar asesoramiento genético a los padres de los pacientes con craneosinostosis no sindrómica con la finalidad de que comprendan los aspectos médicos de la enfermedad, la etiología, el curso probable de la misma, el manejo posible, así como el riesgo de recurrencia para una correcta toma de decisiones.
Evidencia: en un estudio de serie de casos se encontró que los niños con diagnóstico clínico de craneosinostosis no sindrómica presentaron más alteraciones genéticas como causa de la sinostosis si esta era unicoronal o bicoronal (10 de 48 pacientes) que en los casos de sinostosis sagital o metópica (ninguno de 55) (p=0.0003). Por otra parte, se observó que niños con mutaciones en los genes FGFR2 exones iiia/iiic, FGFR3 (P250R) y TWIST1 presentan complicaciones quirúrgicas más graves que los que no tienen una causa genética (p<0.05)28. En otro estudio de serie de casos que incluyó 182 pacientes con craneosinostosis, se les realizó análisis molecular de los genes FGFR1, FGFR2, FGFR3, TWIST1, EFNB1 y TCF12, encontrando alteraciones genéticas en 119 (65%)29. En una revisión sistemática se encontró una concordancia en gemelos monocigóticos con craneosinostosis del 60.9% comparado con gemelos dicigóticos, en la que fue del 5.3% (p<0.0001)30.
Recomendación: se recomienda realizar tratamiento quirúrgico antes de los 12 meses de edad en pacientes con craneosinostosis. Muy baja calidad de evidencia/Recomendación fuerte.
Evidencia: se encontraron 3 estudios que compararon el cociente intelectual de pacientes con craneosinostosis que recibieron tratamiento quirúrgico antes del año de edad frente a pacientes operados después del año de edad, encontrando diferencia estadísticamente significativa en el cociente intelectual, siendo mayor en aquellos que fueron operados antes del año de edad: Arnaud et al. (p=0.056), Bottero et al. (p=0.023) y Patel et al. (p<0.01)31–33. Sin embargo, debido a la heterogeneidad en sus diseños no fue posible realizar un metaanálisis. Un estudio retrospectivo que incluyó 796 pacientes con craneosinostosis y tratamiento quirúrgico, cuyo objetivo fue determinar la incidencia de complicaciones, reintervenciones y factores predictivos, reportó34:
Factores predictivos de complicaciones quirúrgicas:
- •
Tratamiento quirúrgico realizado antes de los 9 meses de edad (OR 1.88, IC 95% 1.01-3.51, p=0.046).
- •
Sinostosis múltiple (OR 2.18, IC 95% 1.13-4.20, p=0.019).
- •
Craneosinostosis sindrómica (OR 2.36, IC 95% 1.31-4.25, p=0.004).
- •
Menor edad en el momento de la cirugía (OR 1.05, IC 95% 1.02-1.08, p=0.001).
- •
Mayor tiempo de duración de la cirugía (OR 1.44, IC 95% 1.24-1.86, p<0.001).
- •
Mayor cantidad de transfusión sanguínea (OR 2.97, IC 95% 1.65-5.35, p<0.001).
- •
Pacientes a quienes se les realizó craneoplastia tipo Spring (OR 4.40, IC 95% 1.32-14.63, p=0.016).
Los factores asociados a un mayor riesgo de sinostosis recurrente fueron:
- •
Craneosinostosis sindrómica (p<0.001).
- •
Craneosinostosis múltiple (p<0.001).
Los factores que se asociaron a la posibilidad de requerir un procedimiento quirúrgico adicional fueron:
- •
Pacientes operados antes de los 9 meses de edad (OR 13.06, IC 95% 2.12-80.24, p<0.006).
- •
Sinostosis múltiple (OR 11.93, IC 95% 4.07-34.94, p<0.001).
- •
Craneosinostosis sindrómica (OR 7.23, IC 95% 2.75-19.00, p<0.001).
Recomendación: se recomienda realizar el tratamiento quirúrgico de acuerdo con el tipo de craneosinostosis. Muy baja calidad de evidencia/Recomendación fuerte.
Evidencia: en un estudio retrospectivo se evaluaron los resultados y las complicaciones del tratamiento quirúrgico de craneosinostosis en 283 pacientes tratados con 12 tipos de técnicas quirúrgicas, encontrando lo siguiente35:
Las complicaciones más frecuentes en el postoperatorio fueron:
- •
Hipertermia de origen indeterminado (13.43%).
- •
Infección (7.5%).
- •
Hematoma subcutáneo (5.3%).
- •
Desgarros durales (5%).
- •
Fugas de líquido cefalorraquídeo (2.5%).
El número de complicaciones fue mayor en el grupo de pacientes reoperados (12.8% del total). En el subconjunto de reintervenciones, la infección se presentó en el 62.5%, los desgarros durales en el 93% y las fugas de líquido cefalorraquídeo en el 75% del total.
En su estudio retrospectivo, que incluyó a 796 pacientes con craneosinostosis, Lee et al.34 reportaron:
- •
La remodelación de la bóveda craneal total conlleva un mayor riesgo de complicaciones (16.3%) en comparación con la remodelación sencilla (6.54%) y la craniectomía extendida (5.41%) (OR 3.4, IC 95% 1.1-10.8, p=0.037).
- •
La fractura dural se asoció con un mayor riesgo de fuga de líquido cefalorraquídeo (OR 18.9, IC 95% 3.9-92.5, p<0.001).
- •
Se identificó como predictor de complicaciones quirúrgicas la realización de craneoplastia tipo Spring (OR 4.40, IC 95% 1.32-14.63, p=0.016).
En su estudio retrospectivo, Kim et al. compararon la cirugía de remodelación craneal con la cirugía con colocación de distractores en pacientes con craneosinostosis, encontrando diferencias estadísticamente significativas a favor de la técnica con distractores solo en la pérdida sanguínea (p=0.002) y el volumen requerido de transfusión sanguínea (p=0.001)36.
Recomendación: se recomienda realizar suturectomía y remodelación craneal o distractores tipo Spring en pacientes con escafocefalia. Muy baja calidad de evidencia/Recomendación débil.
Evidencia: una revisión sistemática que comparó la remodelación total de la bóveda, la craneoplastia tipo Spring y la suturectomía para el tratamiento de la escafocefalia por medio del índice cefálico no encontró diferencia estadísticamente significativa de la remodelación craneal comparada con la técnica tipo Spring, pero reportó una pequeña diferencia estadísticamente significativa a favor de la remodelación craneal comparada con la suturectomía abierta (DME 1.47, IC 95% 0.47-2.48). Sin embargo, como existen diferentes técnicas quirúrgicas y desenlaces que no se incluyeron en esta revisión, se realizó una nueva para encontrar otros estudios de intervención en escafocefalia37.
En su serie de casos de 75 pacientes con escafocefalia, David et al. compararon la colocación de distractores tipo Spring con la cirugía de remodelación craneal y encontraron mejores resultados en la técnica con distractores tipo Spring, tales como38:
- •
Menor pérdida sanguínea (25 frente a 255ml).
- •
Menor media de transfusión sanguínea (0 frente a 356ml).
- •
Menos días de estancia en la Unidad de Cuidados Intensivos (0 frente a 18 días).
- •
Menor estancia media hospitalaria (22.7 frente a 94.2 días).
- •
Menor costo en dólares (8,339.32 frente a 27,212.49$).
Un estudio retrospectivo comparó a pacientes con escafocefalia tratados con craneoplastia tipo Spring frente a pacientes tratados con osteotomía en barril; encontró diferencias estadísticamente significativas a favor de la craneoplastia tipo Spring con respecto a: pérdida sanguínea (p=0.001), menor estancia hospitalaria (p=0.009) y menor tiempo quirúrgico (p=0.002). No encontró diferencias en el índice craneal posquirúrgico entre ambos procedimientos39. Un estudio prospectivo comparó a pacientes que presentaban escafocefalia tratados antes de los 6 meses de edad con craneotomía endoscópica frente a aquellos tratados con craneoplastia total de la bóveda; encontró que los pacientes tratados con esta última tenían mayor cociente intelectual (p<0.05)40. En la escafocefalia y la sinostosis de múltiples suturas se produjo el mayor número de complicaciones con la remodelación completa de la bóveda craneal (desmantelamiento holocraneal), seguida de las técnicas de distracción frontoorbitaria35.
Recomendación: no existe suficiente evidencia para recomendar una técnica quirúrgica para pacientes con trigonocefalia y plagiocefalia anterior. Muy baja calidad de evidencia/Sin recomendación.
Punto de buena práctica: se aconseja realizar la técnica de avance frontoorbitario para pacientes con trigonocefalia y plagiocefalia anterior.
Evidencia: un estudio retrospectivo reportó que en sinostosis de múltiples suturas, trigonocefalia y plagiocefalia, la osteotomía asistida por endoscopia presentó la menor tasa de complicaciones, seguida por el avance frontoorbitario. No se encontraron otros ensayos clínicos o estudios cuasiexperimentales bien diseñados que compararan alguna de las técnicas quirúrgicas para trigonocefalia y plagiocefalia35.
Punto de buena práctica: todo paciente con sospecha de craneosinostosis no sindrómica debe referirse a Neurocirugía o Cirugía Plástica para confirmación diagnóstica y tratamiento.
Punto de buena práctica: todo paciente con diagnóstico de craneosinostosis no sindrómica se debe enviar a Genética para determinar la etiología y brindar asesoramiento genético.
Punto de buena práctica: todo paciente con diagnóstico de craneosinostosis no sindrómica se debe enviar a Rehabilitación para vigilancia del neurodesarrollo e intervención temprana para evitar secuelas neurológicas.
Recomendaciones clave- •
Se recomienda realizar una exploración física dirigida para descartar craneosinostosis no sindrómica a los recién nacidos con factores de riesgo.
- •
Se recomienda realizar radiografía de cráneo (anteroposterior y lateral) y tomografía craneal con reconstrucción 3D en pacientes con sospecha de craneosinostosis.
- •
Se recomienda que a los niños con sinostosis bicoronal o unicoronal confirmada se les realice un estudio genético molecular que incluya las mutaciones en FGFR2 exones iiia/iiic, FGFR3 (P250R) y TWIST1 para determinar la etiología.
- •
Se recomienda realizar tratamiento quirúrgico antes de los 12 meses de edad en pacientes con craneosinostosis.
- •
Se aconseja realizar suturectomía y remodelación craneal o distractores tipo Spring en pacientes con escafocefalia.
- •
Todo paciente con sospecha de craneosinostosis no sindrómica debe referirse a Neurocirugía o Cirugía Plástica para confirmación diagnóstica y tratamiento.
- •
Todo paciente con diagnóstico de craneosinostosis no sindrómica se debe enviar a Genética para determinar la etiología y brindar asesoramiento genético.
- •
Todo paciente con diagnóstico de craneosinostosis no sindrómica se debe enviar a Rehabilitación para vigilancia del neurodesarrollo e intervención temprana para evitar secuelas neurológicas.
Se emitieron 7 recomendaciones y 8 puntos de buena práctica en esta guía, los cuales servirán como apoyo para la toma de decisiones en la atención integral de los pacientes con craneosinostosis.
NotaPara mayor información y solicitud de anexos sobre la integración de la guía de práctica clínica para el diagnóstico, tratamiento y rehabilitación de craneosinostosis no sindrómica en los 3 niveles de atención se sugiere consultar el sitio web del Catálogo Maestro de Guías de Práctica Clínica del CENETEC (http://www.cenetec.salud.gob.mx/contenidos/gpc/catalogoMaestroGPC.html).
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesEsta guía fue elaborada con financiamiento de la Secretaría de Salud del Estado de Puebla, el Hospital General de Puebla Eduardo Vázquez Navarro, el Hospital para el Niño Poblano y el Centro de Rehabilitación Infantil Teletón Puebla. Los puntos de vista de la entidad financiadora no han influido en el contenido de la guía. Se han registrado y abordado los conflictos de intereses de los miembros del grupo elaborador de la guía mediante una carta de declaración de no conflicto de interés.
Se agradece a las autoridades del Hospital General de Puebla Dr. Eduardo Vázquez Navarro, Hospital para el Niño Poblano y al Centro de Rehabilitación Infantil Teletón Puebla las gestiones realizadas para que el personal adscrito al centro o grupo de trabajo que desarrolló la presente guía asistiera a los eventos de capacitación en Medicina Basada en la Evidencia y temas afines, coordinados por la Secretaría de Salud, y el apoyo, en general, al trabajo de los autores.
Asimismo, se agradece a las autoridades del Instituto Nacional de Pediatría que participó en los procesos de validación y verificación su valiosa colaboración en esta guía.