




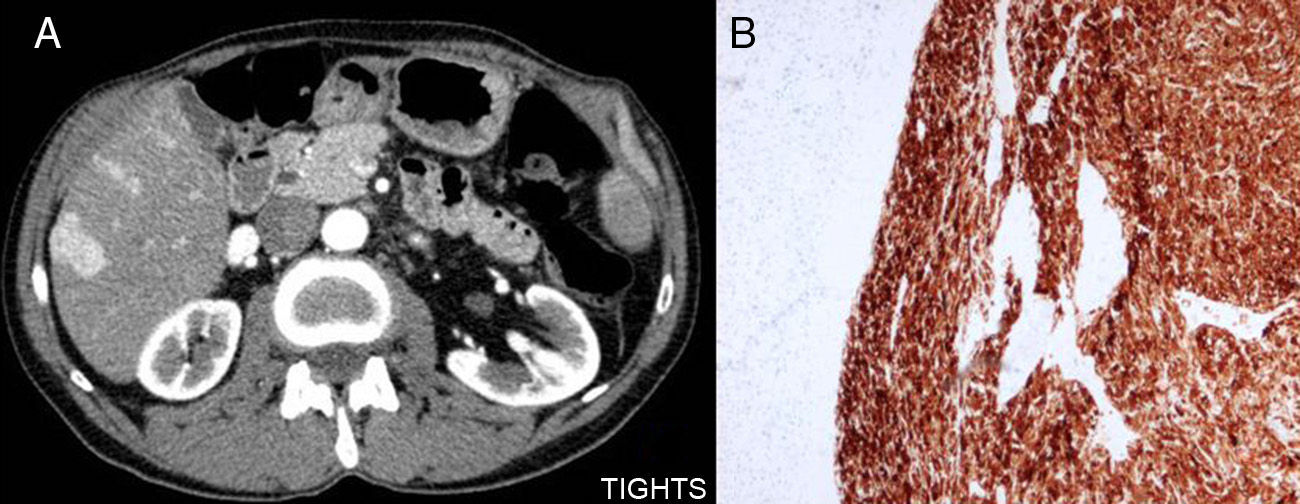
We present the case of a 62-year-old male patient, with no prior pathologic history, who, in 2004, came to our consultation with a palpable abdominal tumour in the left flank that was not painful and adhered to deep planes. CT scan showed a mass that was probably retroperitoneal in origin and had a myxoid appearance. In addition, a liver nodule was detected, which was probably metastatic (Fig. 1).
 Residual pancreas and liver metastases and (B) tumour staining with melanin.")
We decided to perform en bloc surgical resection followed by resection of the tail of the pancreas and metastasectomy of the concomitant liver lesion. The definitive pathology study of the tumour demonstrated that it was compatible with malignant angiomyolipoma, as was the resected liver nodule. Immunohistochemistry showed positive results for muscle markers such as polyactin and vimentin, as well as melanocytic markers like HMB-45 and melan-A, compatible with malignant angiomyolipoma; these immunohistochemistry characteristics are also shared with perivascular epithelioid cell tumours (PEComas) (Fig. 1).
The patient underwent adjuvant chemotherapy but received only 2 cycles of adriamycin+ifosfamide due to toxicity and gastrointestinal bleeding. On follow-up CT scans, multiple pulmonary and hepatic metastases were observed. These lesions remained stable until December 2011, when the CT scan showed that one of the liver lesions, situated in segment II, had increased in size.
After considering the stability of the remaining metastatic lesions and the good condition of the patient, the Oncologic Committee decided to perform another surgery to resect the growing liver metastasis, plus intraoperative radiofrequency ablation of the other macroscopically visible lesions. We completed liver segmentectomy of segments ii and iii, and radiofrequency ablation of 5 metastatic liver tumours. Once again, the pathology study reported the 6cm mass to be compatible with a PEComa. The patient was discharged on the 6th day post-op without complications and has remained stable with no radiological evidence of disease progression.
In the review of the literature motivated by this case, we found that PEComa tumours were described for the first time in 1943 by Apitz et al.1 Later, immunohistological studies, such as the one by Zamboni et al. in 1996,2 demonstrated their positivity for HMB-45 melanocytic marker, imitating pulmonary sugar cell tumours and angiomyolipomas of the kidney and liver. In light of these findings, they suggested a relationship between these tumours and proposed the name PEComas due to the epithelioid morphology of the cells observed in pathology studies.
The World Health Organization (WHO) defines PEComas as “mesenchymal tumours composed of histologically and immunohistochemically distinctive perivascular epithelioid cells”.3
PEComas are formed by round, well-defined cells that are arranged in an epithelioid manner, with a granular, eosinophilic cytoplasm, inner glycogen and marked nuclear pleomorphism.4 In all cases, their location is in blood vessel walls, and they are also composed of a fine capsule.5
Immunohistochemistry studies show PEComas are positive for melanocytic markers, such as HMB-45 or melan-A, as well as muscle markers, like actin or desmin.4
PEComas are extremely rare tumours that, due to their perivascular disposition, can be found in almost any part of the body, and there are reports in the literature in several parts of the organism; meanwhile, they can affect patients of any age and either sex.6–9 As for distribution, there was a greater predisposition towards females, although this fact had not been confirmed statistically. In our review of the literature, we made an approximate statistical assessment, after which we are able to confirm that these tumours show a predilection for females (P<.05).
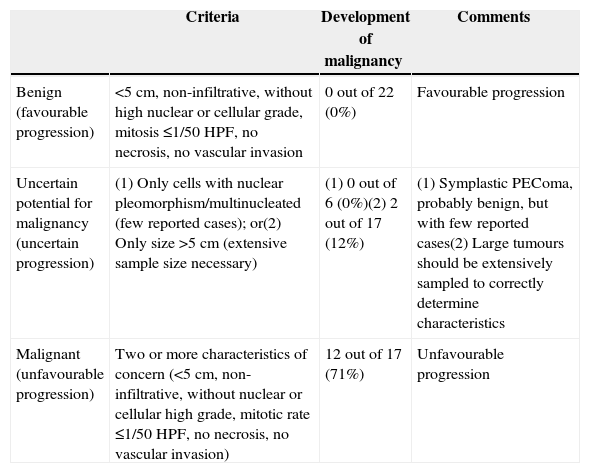
Initially, these tumours were considered benign. However, as our understanding of them has advanced, multiple cases have been reported in which there was already local dissemination or metastasis at the time of diagnosis.5 Folpe et al. observed that the most influential factors in the prognosis of PEComas are: size >5cm, multiple nuclei in each cell, the degree of cellularity in the tumour, high mitotic rate and presence of necrosis and vascular invasion (Table 1).
Proposed Classification for PEComas.
| Criteria | Development of malignancy | Comments | |
|---|---|---|---|
| Benign (favourable progression) | <5cm, non-infiltrative, without high nuclear or cellular grade, mitosis ≤1/50 HPF, no necrosis, no vascular invasion | 0 out of 22 (0%) | Favourable progression |
| Uncertain potential for malignancy (uncertain progression) | (1) Only cells with nuclear pleomorphism/multinucleated (few reported cases); or(2) Only size >5cm (extensive sample size necessary) | (1) 0 out of 6 (0%)(2) 2 out of 17 (12%) | (1) Symplastic PEComa, probably benign, but with few reported cases(2) Large tumours should be extensively sampled to correctly determine characteristics |
| Malignant (unfavourable progression) | Two or more characteristics of concern (<5cm, non-infiltrative, without nuclear or cellular high grade, mitotic rate ≤1/50 HPF, no necrosis, no vascular invasion) | 12 out of 17 (71%) | Unfavourable progression |
Source: Folpe et al.5
As for treatment, we should mention the limited information available about the response of these tumours to chemotherapy and radiotherapy. In our case, although the patient only received 2 cycles of chemotherapy, this dose was sufficient to avoid progression of the tumour and maintain the patient in partial tumour remission. The lack of consensus about the response of this tumour to radiotherapy led us to decide against this treatment.
Currently, the only treatment that seems to improve survival in these patients is surgery, which has even been curative (to date) in some cases presented in the literature.10 R0 surgical resection is able to maintain certain patients with no tumour recurrence for several years. Likewise, cytoreductive surgery is able to prolong the survival of these patients, as occurred with our patient.
FundingWe, the authors, declare that we have received no funding of any sort.
Please cite this article as: Cuevas Herreros Ó, Escobar Lezcano L, Rodriguez Blaco M, Artigas Raventós V. PEComa, un raro tumor de células epiteloides. Cir Esp. 2015;93:e65–e67.
The information of this manuscript has not been reported at a congress or published in any journal or other scientific media.
recomendados

Cirugía Española (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas