






La pancreatitis autoinmune (PAI) es una enfermedad fibroinflamatoria benigna del páncreas, se manifiesta frecuentemente como ictericia obstructiva asociada a masa pancreática o lesión obstructiva de la vía biliar y presenta una respuesta excelente a corticoides. Aunque no existen estudios a nivel mundial que definan su epidemiología, la PAI se considera una entidad poco frecuente, con una prevalencia estimada del 2% de los pacientes con pancreatitis crónica. Su frecuente presentación clínica y radiológica en forma de masa pancreática e ictericia similar al cáncer de páncreas y la falta de elementos diagnósticos específicos son causa de un elevado porcentaje de resecciones quirúrgicas pancreáticas por una enfermedad benigna que responde a tratamiento médico. En esta revisión detallamos los acuerdos actuales para el diagnóstico, clasificación y tratamiento de la PAI, enfatizando en las series quirúrgicas y en estrategias para mejorar el diagnóstico diferencial con el cáncer de páncreas y evitar así resecciones pancreáticas innecesarias.
Autoimmune pancreatitis (AIP) is defined as a particular form of pancreatitis that often manifests as obstructive jaundice associated with a pancreatic mass or an obstructive bile duct lesion, and that has an excellent response to corticosteroid treatment. The prevalence of AIP worldwide is unknown, and it is considered as a rare entity. The clinical and radiological presentation of AIP can mimic bilio-pancreatic cancer, presenting difficulties for diagnosis and obliging the surgeon to balance decision-making between the potential risk presented by the misdiagnosis of a deadly disease against the desire to avoid unnecessary major surgery for a disease that responds effectively to corticosteroid treatment. In this review we detail the current and critical points for the diagnosis, classification and treatment for AIP, with a special emphasis on surgical series and the methods to differentiate between this pathology and bilio-pancreatic cancer.
La pancreatitis autoinmune (PAI) es una enfermedad fibroinflamatoria benigna del páncreas descrita por primera vez en 1961 como un caso de pancreatitis asociada a hipergammaglobulinemia1. En 1995, Yoshida et al. propusieron el concepto de PAI2. Recientemente, la Asociación Internacional de Pancreatología definió la PAI como una forma particular de pancreatitis que a menudo se manifiesta como ictericia obstructiva asociada o no a masa pancreática, que cursa con cambios histológicos característicos consistentes en infiltrado linfoplasmocitario y fibrosis, y que presenta una respuesta excelente al tratamiento con corticoides3.
Tipos de pancreatitis autoinmuneEl análisis histopatológico del páncreas define 2 patrones con características diferenciales: 1) la pancreatitis esclerosante linfoplasmocitaria (PELP) o PAI sin lesiones epiteliales granulocíticas, y 2) la pancreatitis idiopática ductocéntrica (PIDC) o PAI con lesiones epiteliales granulocíticas. Sin embargo, dado que no siempre es posible disponer de la descripción histológica, se han introducido los términos PAI tipo 1 y tipo 2 con el objetivo de describir las manifestaciones clínicas asociadas a la PELP o la PIDC, respectivamente4.
La pancreatitis tipo 1 es la forma predominante en países asiáticos. Se manifiesta más frecuentemente en hombres (3-4:1), con un pico de presentación durante la sexta década de la vida, puede cursar con elevación de la inmunoglobulina G tipo 4 (IgG4) en suero y se asocia a menudo con afectación fibroinflamatoria de otros órganos. En los pacientes con PAI tipo 1 es característica la resolución de las manifestaciones pancreáticas y extrapancreáticas con corticoides, aunque la recurrencia tras cesar el tratamiento es frecuente, en especial en los casos que cursan con afectación extrapancreática5.
La pancreatitis tipo 2 se describe más en Europa y Estados Unidos. Afecta por lo general a pacientes más jóvenes (una década antes que la PAI tipo 1), sin predilección por el sexo, no cursa con elevación sérica de IgG4, no se asocia afectación de otros órganos y, en una elevada proporción de pacientes (11-30%), existe enfermedad inflamatoria intestinal asociada (colitis ulcerosa más frecuente que enfermedad de Crohn). La respuesta al tratamiento con corticoides es buena y las recaídas son infrecuentes. Dado que la PAI tipo 2 carece de marcadores serológicos (no aumento de IgG4) y de afectación de otros órganos, su diagnóstico definitivo requiere estudio histológico del páncreas. Ello explica en parte que la PAI tipo 2 sea diagnosticada con menos frecuencia que la tipo 15.
Manifestaciones clínicasLa manifestación más frecuente es la ictericia obstructiva causada por una masa en la cabeza pancreática (hasta en el 59% de los casos) o por engrosamiento de la pared del colédoco6. También se puede manifestar en forma de pancreatitis aguda única o recurrente o evolucionar a pancreatitis crónica con calcificaciones e insuficiencia pancreática exocrina y endocrina7. Los síntomas relacionados con la afectación extrapancreática son otra forma de presentación, por ejemplo, la tumoración lagrimal o salival, tos o disnea por lesiones pulmonares o lumbago secundario a fibrosis retroperitoneal o hidronefrosis6.
Cambios histopatológicosLa PAI presenta unos cambios histopatológicos en el páncreas, bien definidos, que son fácilmente distinguibles de los cambios ocurridos en otros tipos de pancreatitis (crónica alcohólica u obstructiva). Algunos de estos son hallazgos comunes al tipo 1 y tipo 2 y otros sirven para distinguir entre ambos tipos8,9.
Hallazgos histopatológicos comunes a la pancreatitis autoinmune tipo 1 y tipo 2La infiltración linfoplasmocitaria y el estroma celular inflamatorio son hallazgos muy característicos de la PAI8. El infiltrado linfoplasmocitario es denso y se acentúa en torno a los ductos de mediano y gran tamaño, comprimiendo la luz ductal (imagen ductal en herradura o en estrella muy característica de la PAI) que difiere de la dilatación ductal (característica de la pancreatitis crónica de otro origen). La infiltración linfoplasmocitaria se extiende de forma difusa por el parénquima pancreático, donde se acompaña de fibrosis y atrofia acinar. El resultado es un estroma celular inflamatorio, en el cual abundan los linfocitos, las células plasmáticas y áreas parcheadas de eosinófilos, propios de la PAI pero no de otros tipos de pancreatitis crónica.
Hallazgos característicos de la pancreatitis autoinmune tipo 1La fibrosis estoriforme, la flebitis obliterativa, los folículos linfoides prominentes y las células plasmáticas IgG4+ son hallazgos muy característicos de la PAI tipo 1, aunque también se observan en menor proporción en la tipo 29. La fibrosis estoriforme, o arremolinada, es un tipo peculiar de fibrosis ocasionado por un entramado de fibras cortas de colágeno entrelazadas en diversas direcciones e infiltradas por un denso componente linfoplasmocitario. Este patrón se describe en el 90% de las PAI tipo 1 y el 29% de las PAI tipo 2. La flebitis obliterativa traduce la inflamación de las venas por infiltración linfoplasmocitaria y consiguiente obstrucción de la luz vascular. Si bien es difícil de reconocer, su identificación es de gran interés por ser un signo patognomónico de PAI. Esta alteración se describe en el 90% de la PAI tipo 1 y en el 57% de la PAI tipo 2. La existencia de agregados y folículos linfoides prominentes en el parénquima y grasa peripancreática es otro hecho característico de la PAI (100% en la tipo 1 y 47% en la tipo 2), pero que también se observa en aproximadamente la mitad de casos de pancreatitis crónica alcohólica y pancreatitis crónica obstructiva. La detección de abundantes células plasmáticas IgG4 (> 10 células/campo de gran aumento [CGA]) es un dato clave en el diagnóstico de PAI tipo 1, en tanto que en la PAI tipo 2 no existen células plasmáticas IgG4 o son poco abundantes (< 10/CGA). Es importante considerar que estas células también pueden observarse en otras formas de pancreatitis crónica (11-57%) y en el adenocarcinoma ductal de páncreas (12-47%).
Hallazgo característico de la pancreatitis autoinmune tipo 2Las lesiones epiteliales granulocíticas son patognomónicas de la PAI tipo 29. Estas lesiones consisten en infiltrados de neutrófilos que afectan a los ductos de mediano y pequeño tamaño así como a las células acinares, ocasionando la destrucción celular y la obliteración de la luz.
Pruebas de imagenPara el diagnóstico de PAI debemos buscar cambios parenquimatosos y ductales característicos, aunque en muchas ocasiones pueden ser indistinguibles de los presentes en el cáncer de páncreas6. La imagen típica es un páncreas aumentado de tamaño de forma difusa («páncreas en salchicha») con pérdida de la lobularidad. En el estudio por tomografía axial computarizada (TC) es característica la hipoatenuación del parénquima en la fase pancreática y el realce tardío durante la fase venosa. En la resonancia magnética (RM) es también característico un aumento del tamaño del páncreas, hipointenso en secuencias potenciadas en T1 en comparación con el páncreas no afecto o con el hígado, hiperintenso en secuencias potenciadas en T2, y con realce tardío en fase venosa. Es un hallazgo típico de la PAI el halo periférico hipoatenuado en la TC con contraste e hipointenso en las imágenes T1 y T2 de la RM. Sin embargo, la PAI puede manifestarse también como una masa focal pancreática hipodensa o hipointensa en la TC y RM, respectivamente, y es en estos casos cuando el diagnóstico diferencial con el cáncer de páncreas es más complicado (fig. 1). La wirsungrafía mediante RM o colangiografía retrógrada endoscópica pueden aportar información clave respecto a los cambios ductales. Es característico de la PAI la estenosis larga (> 1/3 del conducto del Wirsung), estenosis múltiples o estenosis focales, todo ello sin dilatación proximal10. La ecoendoscopia es otra prueba de gran valor en el diagnóstico de la PAI especialmente por la posibilidad que ofrece de conseguir citología o biopsias pancreáticas para estudio anatomopatológico3,11,12.
 en forma de masa pancreática asociada a ictericia indolora. A) Ecografía abdominal que muestra una zona focal hipoecoica en la cabeza del páncreas con relación al resto de la glándula. B) La secuencia potenciada en T2 del examen de resonancia magnética muestra una zona focal con aumento de la señal en la misma zona del páncreas, sin clara distorsión de los contornos ni aumento del tamaño de la glándula. C) En las secuencias colangiográficas se puede apreciar una ligera estenosis focal en el tamaño del conducto pancreático, que permanece permeable, y que no condiciona dilatación proximal. D) La zona focal del páncreas aparece menos realzada que el parénquima vecino en la fase arterial del estudio dinámico; E) presenta mayor realce en fases tardías.")
Imágenes radiológicas de un paciente portador de pancreatitits autoinmune (PAI) en forma de masa pancreática asociada a ictericia indolora. A) Ecografía abdominal que muestra una zona focal hipoecoica en la cabeza del páncreas con relación al resto de la glándula. B) La secuencia potenciada en T2 del examen de resonancia magnética muestra una zona focal con aumento de la señal en la misma zona del páncreas, sin clara distorsión de los contornos ni aumento del tamaño de la glándula. C) En las secuencias colangiográficas se puede apreciar una ligera estenosis focal en el tamaño del conducto pancreático, que permanece permeable, y que no condiciona dilatación proximal. D) La zona focal del páncreas aparece menos realzada que el parénquima vecino en la fase arterial del estudio dinámico; E) presenta mayor realce en fases tardías.
Actualmente carecemos de marcadores serológicos específicos para el diagnóstico de PAI. La elevación de los niveles séricos de IgG4 es un dato característico de la PAI tipo 1, aunque su interpretación merece algunas consideraciones en cuanto a su sensibilidad y especificidad diagnóstica13. En cuanto a la sensibilidad, la PAI tipo 2 nunca cursa con aumento de IgG4 y, entre los pacientes con PAI tipo 1, un porcentaje de ellos son seronegativos para IgG414. En referencia a su especificidad, esta viene determinada en gran parte por el nivel de corte establecido, y es importante considerar que la IgG4 puede hallarse elevada en otras enfermedades pancreáticas (especialmente en el cáncer de páncreas y pancreatitis crónica) y extrapancreáticas (dermatitis atópica, asma, pénfigo, parasitosis). Si se consideran positivos valores por encima del nivel superior de la normalidad (IgG4>135mg/dl), la sensibilidad de la IgG4 es limitada (79-93%)15,16, siendo positivos el 5% de los sujetos controles y el 10% de los pacientes con cáncer de páncreas15. Si el valor de corte se establece para aquellos superiores a 2 veces la normalidad (IgG4>280mg/dl), ningún control y solo el 1% de los pacientes con cáncer de páncreas muestran IgG4 elevada, siendo en estos casos la especificidad de este marcador del 99%15. Por el momento no disponemos de datos epidemiológicos que definan la precisión diagnóstica de la IgG4 en España, y los datos procedentes de diversos estudios son muy heterogéneos según el área geográfica (mucho más prevalente en países asiáticos que en Europa), el nivel de corte o el tipo de PAI analizada13. Otros parámetros que pueden acompañar a la PAI son la hipergammaglobulinemia, la elevación de la IgG, la hipereosinofilia o la existencia de anticuerpos antinucleares y del factor reumatoide. Si bien estos parámetros pueden ayudar a confirmar el diagnóstico, ninguno de ellos ha sido reconocido dentro de los criterios diagnósticos establecidos por los criterios internacionales para el diagnóstico de PAI3.
Afectacion de otros órganosEl 50-70% de pacientes con PAI tipo 1 presentan afectación de otros órganos como parte de una enfermedad sistémica asociada a IgG43,17,18. Las manifestaciones extrapancreáticas pueden preceder, ser simultáneas o ser posteriores a la PAI. El diagnóstico de afectación de otros órganos se puede realizar a partir de la afectación histológica (infiltración de células plasmáticas IgG4 del el tejido afecto), imagen (estenosis del colédoco proximal, fibrosis retroperitoneal), examen clínico (aumento del tamaño de las glándulas salivales) y respuesta a esteroides18,19. Hay gran número de tejidos que pueden estar afectados, siendo la afectación más frecuente (50-90% de casos) la del árbol biliar (colangitis esclerosante asociada a IgG4), la cual se manifiesta habitualmente como ictericia obstructiva. Otras localizaciones posibles son los ganglios linfáticos, glándulas salivales y lacrimales, tiroides, retroperitoneo, vesícula biliar, hígado, aorta, riñones y uréter, mama, pulmón, sistema nervioso central y próstata. Una característica que comparten los diferentes órganos afectados en el contexto de síndrome asociado a IgG4 es la tendencia a formar lesiones tumefactas o pseudotumores, la existencia de lesiones histológicas similares a las halladas en el páncreas y la elevación, aunque no siempre, de la IgG4 en suero. Otros trastornos autoinmunes (p.ej. artritis reumatoide, psoriasis, síndrome de Sjögren) no se consideran afectación de otros órganos en el diagnóstico de PAI19.
TratamientoEl objetivo del tratamiento es conseguir la desaparición de los síntomas y la resolución de las manifestaciones pancreáticas y extrapancreáticas observadas en las pruebas de imagen20. El tratamiento de elección son los corticoides, dada su buena respuesta tanto en la PAI tipo 1 como en la tipo 2, aunque la recurrencia tras el cese del tratamiento es elevada, especialmente en la PAI tipo 121. Aunque no existe un protocolo terapéutico estandarizado, la mayoría de pautas recomiendan prednisona a una dosis inicial de 35-40mg/día22, o 0,6-1mg/kg/día según el consenso internacional para el diagnóstico de PAI3, durante 4 semanas y, si ha habido respuesta radiológica y clínica, disminuir gradualmente la dosis a lo largo de 3-4 meses. Algunos grupos recomiendan mantener el tratamiento con corticoides a dosis bajas (2,5-5mg/día) durante 3 años en la PAI tipo 1, dada su elevada tasa de recurrencia23. La reintroducción de los corticoides o el inicio de inmunosupresores como la azatioprina, el metotrexato y el micofenolato y el rituximab son alternativas terapéuticas utilizadas en caso de recurrencia al terminar el tratamiento con esteroides20,24. Se consideran factores predictores de recurrencia la estenosis proximal del conducto biliar y la persistencia de elevación de IgG420. Algunos casos, especialmente en la PAI tipo 2, presentan remisión espontánea sin tratamiento esteroideo25.
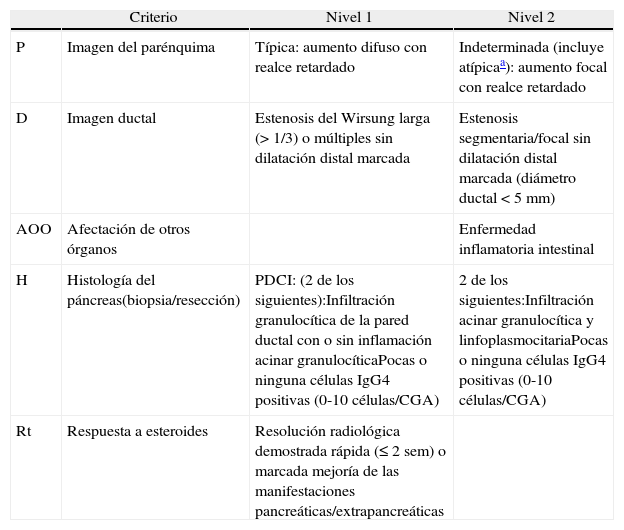
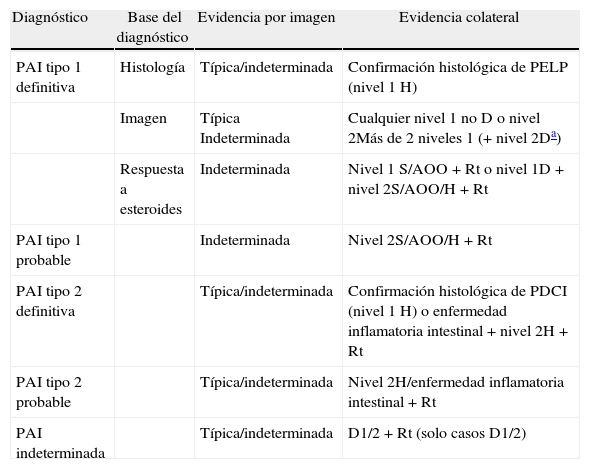
DiagnósticoLa Asociación Internacional de Pancreatología elaboró en 2010 el Consenso Internacional sobre Criterios Diagnósticos (CICD) de la PAI3. Este documento unifica los criterios diagnósticos definidos hasta entonces por diversas sociedades (japonesa, coreana, asiática, Clínica Mayo, Mannheim, italiana). El CICD establece el diagnóstico de PAI mediante la combinación de uno o más de los siguientes aspectos: 1) Hallazgos en la imagen: a) del parénquima pancreático (mediante TC o RM) y b) del conducto pancreático (mediante colangio-RM o colangiografía retrógrada endoscópica); 2) serología (IgG, IgG4, anticuerpos antinucleares); 3) afectación de otros órganos; 4) histología del páncreas y 5) respuesta a corticoides. Cada uno de estos aspectos se categorizan en nivel 1 y nivel 2 según su fiabilidad diagnóstica. Tras aplicar estos criterios se puede llegar al diagnóstico definitivo o probable de PAI tipo 1 (tablas 1 y 2) o tipo 2 (tablas 2 y 3), aunque en algunos casos no es posible distinguir entre ambos tipos (pancreatitis autoinmune indeterminada, tabla 2)3.
Clasificación de los criterios diagnósticos de pancreatitis autoinmune tipo 1 en nivel 1 y 2
| Criterio | Nivel 1 | Nivel 2 | |
| P | Imagen del parénquima | Típica: aumento difuso con realce retardado | Indeterminada (incluye atípicaa): aumento focal con realce retardado |
| D | Imagen ductal | Estenosis del Wirsung larga (> 1/3) o múltiples sin dilatación distal marcada | Estenosis segmentaria/focal sin dilatación distal marcada (diámetro ductal<5mm) |
| S | Serología | IgG4>× 2 sobre valor límite alto normalidad | IgG4, × 1-2 sobre valor límite alto normalidad |
| AOO | Afectación a otros órganos | a o ba) Histología (≥ 3 de los siguientes):Marcada infiltración linfoplasmocitaria sin infiltración granulocíticaFibrosis estoriformeFlebitis obliterativa> 10 células IgG4 positivas/CGAb) Imagen (≥ 1 de los siguientes):Estenosis biliar proximal segmentaria/múltiple (hiliar/intrahepática) o proximal y distalFibrosis retroperitoneal | a o ba) Histología. Incluye biopsia papilla de Vater (ambos de los siguientes):Marcada infiltración linfoplasmocitaria sin infiltración granulocítica10 células IgG4 positivas/CGAb) Evidencia clínica o radiológica (≥ 1 de los siguientes:Aumento simétrico de las glándulas salivales/lacrimalesAfectación radiológica renal descrita en asociación con la PAI |
| H | Histología del páncreas | PELP, biopsia o resección (≥ 3 de los siguientes):Infiltrado linfoplasmocitario periductal sin infiltración granulocíticaFlebitis obliterativaFibrosis estoriforme≥ 10 células IgG4 positivas/CGA | PELP, biopsia (≥ 2 de los siguientes):Infiltrado linfoplasmocitario periductal sin infiltración granulocíticaFlebitis obliterativaFibrosis estoriforme≥ 10 células IgG4 positivas/CGA |
| Rt | Respuesta a esteroides | Resolución radiológica demostrada rápida (≤ 2 semanas) o marcada mejoría de las manifestaciones pancreáticas/extrapancreáticas |
CGA: campo gran aumento; PAI: pancreatitis autoinmune; PELP: pancreatitis esclerosante linfoplasmocitaria.
Atípica: masa de baja densidad, dilatación ductal o atrofia pancreática distal. Estos hallazgos atípicos en un paciente con ictericia obstructiva son muy sugestivos de cáncer de páncreas. Estos casos deben ser considerados como cáncer de páncreas a menos que existan importantes evidencias colaterales de PAI y se haya realizado un exhaustivo diagnóstico para descartar malignidad.
Clasificación de los criterios diagnósticos de pancreatitis autoinmune tipo 2 en nivel 1 y 2
| Criterio | Nivel 1 | Nivel 2 | |
| P | Imagen del parénquima | Típica: aumento difuso con realce retardado | Indeterminada (incluye atípicaa): aumento focal con realce retardado |
| D | Imagen ductal | Estenosis del Wirsung larga (> 1/3) o múltiples sin dilatación distal marcada | Estenosis segmentaria/focal sin dilatación distal marcada (diámetro ductal<5mm) |
| AOO | Afectación de otros órganos | Enfermedad inflamatoria intestinal | |
| H | Histología del páncreas(biopsia/resección) | PDCI: (2 de los siguientes):Infiltración granulocítica de la pared ductal con o sin inflamación acinar granulocíticaPocas o ninguna células IgG4 positivas (0-10 células/CGA) | 2 de los siguientes:Infiltración acinar granulocítica y linfoplasmocitariaPocas o ninguna células IgG4 positivas (0-10 células/CGA) |
| Rt | Respuesta a esteroides | Resolución radiológica demostrada rápida (≤ 2 sem) o marcada mejoría de las manifestaciones pancreáticas/extrapancreáticas |
CGA: campo gran aumento;;PDCI: pancreatitis ductocéntrica idiopática.
Atípica: masa de baja densidad, dilatación ductal o atrofia pancreática distal. Estos hallazgos atípicos en un paciente con ictericia obstructiva son muy sugestivos de cáncer de páncreas. Estos casos deben ser considerados como cáncer de páncreas a menos que existan importantes evidencias colaterales de PAI y se haya realizado un exhaustivo diagnóstico para descartar malignidad.
Diagnóstico de pancreatitis autoinmune tipo 1 y tipo 2 en definitiva o probable, e indeterminada
| Diagnóstico | Base del diagnóstico | Evidencia por imagen | Evidencia colateral |
| PAI tipo 1 definitiva | Histología | Típica/indeterminada | Confirmación histológica de PELP (nivel 1H) |
| Imagen | Típica Indeterminada | Cualquier nivel 1 no D o nivel 2Más de 2 niveles 1 (+ nivel 2Da) | |
| Respuesta a esteroides | Indeterminada | Nivel 1 S/AOO+Rt o nivel 1D+nivel 2S/AOO/H+Rt | |
| PAI tipo 1 probable | Indeterminada | Nivel 2S/AOO/H+Rt | |
| PAI tipo 2 definitiva | Típica/indeterminada | Confirmación histológica de PDCI (nivel 1H) o enfermedad inflamatoria intestinal+nivel 2H+Rt | |
| PAI tipo 2 probable | Típica/indeterminada | Nivel 2H/enfermedad inflamatoria intestinal+Rt | |
| PAI indeterminada | Típica/indeterminada | D1/2+Rt (solo casos D1/2) |
AOO: afectación de otros órganos; D: imagen ductal; H: histología del páncreas; PAI: pancreatitis autoinmune; PDCI: pancreatitis ductocéntrica idiopática; PELP: pancreatitis esclerosante linfoplasmocitaria; Rt: respuesta a esteroides; S: serología.
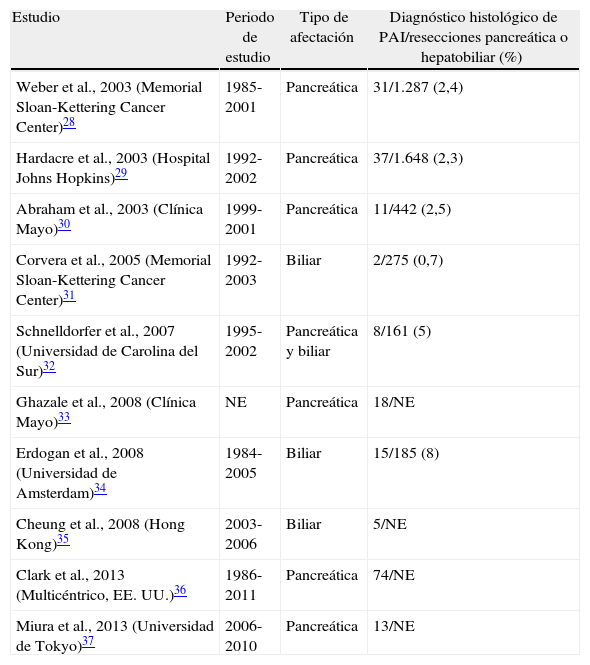
La prevalencia de la PAI tanto en países de Oriente como de Occidente no se conoce. En Japón, se estima una prevalencia de 0,82 por 100.000 habitantes y en Estados Unidos representa el 6% de los pacientes con pancreatitis crónica, lo que sitúa a la PAI como una enfermedad rara (aquellas con prevalencia<5 casos/100.000 habitantes)26,27. Sin embargo, a pesar de su rareza, el diagnóstico diferencial de la PAI con la neoplasia de páncreas o de vías biliares es fundamental, dado que tanto su pronóstico como el tratamiento ofrecido son radicalmente diferentes. La presentación clínica y radiológica a menudo indistinguible de la de una neoplasia biliopancreática y la falta de un marcador diagnóstico definitivo llevan a la infravaloración diagnóstica de esta enfermedad, lo cual comporta, en un número no despreciable de casos, una resección quirúrgica pancreática innecesaria. Los trabajos de series quirúrgicas publicados hasta este momento tienen un gran valor científico, puesto que además de servir como punto de partida para reconocer y describir las lesiones histológicas de la PAI, ponen de manifiesto la dificultad para su diagnóstico y nos muestran la necesidad de tener una mayor conciencia y conocimiento de la existencia de esta enfermedad para poder llegar a su diagnóstico antes de indicar la cirugía (tabla 4)28–37.
Series quirúrgicas de la pancreatitis autoinmune
| Estudio | Periodo de estudio | Tipo de afectación | Diagnóstico histológico de PAI/resecciones pancreática o hepatobiliar (%) |
| Weber et al., 2003 (Memorial Sloan-Kettering Cancer Center)28 | 1985-2001 | Pancreática | 31/1.287 (2,4) |
| Hardacre et al., 2003 (Hospital Johns Hopkins)29 | 1992-2002 | Pancreática | 37/1.648 (2,3) |
| Abraham et al., 2003 (Clínica Mayo)30 | 1999-2001 | Pancreática | 11/442 (2,5) |
| Corvera et al., 2005 (Memorial Sloan-Kettering Cancer Center)31 | 1992-2003 | Biliar | 2/275 (0,7) |
| Schnelldorfer et al., 2007 (Universidad de Carolina del Sur)32 | 1995-2002 | Pancreática y biliar | 8/161 (5) |
| Ghazale et al., 2008 (Clínica Mayo)33 | NE | Pancreática | 18/NE |
| Erdogan et al., 2008 (Universidad de Amsterdam)34 | 1984-2005 | Biliar | 15/185 (8) |
| Cheung et al., 2008 (Hong Kong)35 | 2003-2006 | Biliar | 5/NE |
| Clark et al., 2013 (Multicéntrico, EE. UU.)36 | 1986-2011 | Pancreática | 74/NE |
| Miura et al., 2013 (Universidad de Tokyo)37 | 2006-2010 | Pancreática | 13/NE |
Proporción de pacientes diagnosticados de PAI tras estudio histológico de pieza quirúrgica en pacientes intervenidos por sospecha de neoplasia biliopancreática.
NE: no especificado; PAI: pancreatitis autoinmune.
La manifestación clínica más frecuente del la PAI, en especial en la tipo 1, es la ictericia obstructiva indolora asociada a una masa pancreática (68-84%)28,29,38, siendo además la vía biliar el sitio de afectación extrapancreática más frecuente6,39. Una lesión tipo masa en la cabeza pancreática y la afectación del conducto biliar distal ocasionadas por la PAI producen un cuadro clínico en muchos casos indistinguible del cáncer de páncreas. Incluso cuando existe afectación biliar proximal, la sospecha puede ser la de colangiocarcinoma hiliar34.
Existen 3 grandes series quirúrgicas publicadas que describen la casuística de PAI en un gran número de enfermos con resección pancreática. Estas son la de Weber et al. del Memorial Sloan-Kettering Cancer Center de Nueva York, la de Hardacre et al. del Hospital Johns Hopkins en Baltimore y la de Abraham et al. de la Clínica Mayo en Minnesota (tabla 4)28–30. El estudio de Weber recoge 1.287 resecciones pancreáticas realizadas entre 1985 y 200128. De ellas, 159 (12%) presentaron enfermedad benigna en la evaluación anatomopatológica, de las cuales, 29 fueron pancreatitis autoinmunes y 2 pacientes presentaron pseudotumores secundarios a PAI considerados irresecables. Estos 31 pacientes representan el 2,7% del total de los pacientes operados y el 19,5% de los pacientes operados con enfermedad benigna. En estos 31 pacientes, la edad media fue de 62 años, 68% fueron hombres, 68% presentaron ictericia, 29% dolor abdominal y 19% «enfermedad autoinmune asociada». Las resecciones fueron 23 duodenopancreatectomías cefálicas (DPC), 4 pancreatectomías distales y 2 pancreatectomías totales. Dos pacientes fueron «irresecables» (por infiltración de arteria mesentérica superior y vena porta). Ocho de los 29 pacientes (28%) presentaron «recurrencia» de la enfermedad y 3 de los 4 pacientes con resección distal desarrollaron ictericia posteriormente. Incluso 4 de los 23 pacientes con DPC desarrollaron ictericia después de la resección (3 secundarios a estenosis intrahepáticas múltiples y uno a estenosis de la anastomosis bilioentérica), y una de las 23 DPC desarrolló pancreatitis secundaria a estenosis ductal pancreática28. Este estudio destaca la dificultad de llegar a un diagnóstico correcto de la PAI antes de la cirugía así como que casi un tercio de los pacientes mostraron recurrencia de la enfermedad tras la resección, lo cual advierte de la necesidad de realizar seguimiento posquirúrgico de estos pacientes.
En la casuística del Hospital Johns Hopkins, se revisaron 1.648 resecciones duodenopancreáticas entre 1992 y 200229. De estas, 176 (11%) fueron por pancreatitis crónica, de las cuales 37 casos (21%) correspondieron a PAI. Todos los pacientes con PAI tuvieron sospecha preoperatoria de cáncer de páncreas y todos fueron resecables. La edad media fue de 62 años, 64% fueron hombres, 84% presentaron ictericia, 54% dolor abdominal y 34% asociaron «enfermedad autoinmune». El tipo de intervención fue 26 DPC preservadoras de píloro y 11 DPC clásicas29.
La revisión de Abraham et al. de la Clínica Mayo evalúa 442 piezas de DPC realizadas durante el período de 1999 a 200130. Esta serie identifica 47 piezas (10,6%) negativas para enfermedad neoplásica y de estas, 40 casos fueron intervenidos quirúrgicamente por sospecha de malignidad (9,2%). Las características clínicas que levantaron la sospecha de malignidad en estos 40 casos fueron la lesión tipo masa pancreática en el 67%, la ictericia obstructiva en el 50%, la estenosis de la vía biliar en el 40% y la citología positiva en el 12% de los pacientes. El diagnóstico patológico definitivo de estos 40 pacientes fue el de PAI en 11 (27,5%), pancreatitis crónica asociada al alcohol en 8 (20%), pancreatitis asociada a litiasis biliar en 4 (10%), pancreatitis crónica de etiología indeterminada en 6 (15%), estenosis de la vía biliar aislada de etiología no filiada en 4 pacientes (10%) y colangitis esclerosante en 3 pacientes (7,5%). Este estudio pone de manifiesto que la DPC por enfermedad benigna del páncreas supone un número importante (9,2%) y que de ellas, la PAI representa el diagnóstico más frecuente30.
La experiencia quirúrgica acumulada en pacientes con PAI ha revelado las dificultades técnicas que supone la cirugía pancreática en estos enfermos, que el cirujano debe tener en cuenta para evitar problemas de lesión vascular y hemorragia. Los pacientes con PAI presentan mayor tiempo operatorio y pérdida sanguínea, probablemente secundarios a una disección más difícil de la pieza quirúrgica para la separación de los vasos viscerales, debido a la inflamación peripancreática y a la desfiguración de planos en los tejidos normales28–30,40. Recientemente, Clark et al. han publicado la experiencia quirúrgica a corto y largo plazo de un estudio multicéntrico que incluye pacientes con PAI tratados en la Clínica Mayo (centros de Minnesota y Florida) y en el Hospital General de Massachusetts en el periodo de 1986 a 201136. Este estudio reclutó a 74 pacientes con PAI diagnosticados por histología en la pieza pancreática obtenida tras resección quirúrgica. La edad media de los pacientes fue de 60 años, 69% fueron varones y la indicación quirúrgica fue sospecha de cáncer en el 80% de ellos. El subtipo de PAI se determinó en 63 (85%) pacientes, y de ellos 34 fueron de tipo 1 y 29 de tipo 2. La intervención quirúrgica realizada fue DPC en 56 pacientes (75%), pancreatectomía distal con esplenectomía en 10 pacientes (14%), sin esplenectomía en 5 pacientes (7%) y pancreatectomía total en 3 pacientes (4%). La causa más frecuente de indicación quirúrgica fue la sospecha de neoplasia (n=59, 80%). Este estudio describe dificultad operatoria en 34 pacientes (46%). La reparación/reconstrucción vascular fue necesaria en 15 (20%) pacientes, con una mediana (rango intercuartílico) para tiempo operatorio de 360min (325-415min) y necesidad de transfusión sanguínea en las primeras 24 h postoperatorias en 19 (26%) pacientes. La pérdida sanguínea estimada fue de 600ml (300-1.000ml) y la necesidad de reintervención en los primeros 30 días fue de 4% (3 pacientes). Diez (14%) pacientes presentaron complicaciones mayores, con una muerte perioperatoria (1%) y 2 pacientes con fístulas pancreáticas clínicamente relevantes (grado B/C de la ISGPF). La recurrencia de la sintomatología de la PAI se presentó en el 17% de los pacientes sin afectar su supervivencia global de manera significativa. Estos estudios revelan las dificultades técnicas intraoperatorias y el reto quirúrgico adicional que supone esta afección.
Comentario finalA pesar del creciente conocimiento de la PAI recabado durante los últimos años, la PAI es todavía una enfermedad diagnosticada de forma tardía tras la resección pancreática. Un estudio retrospectivo realizado por Learn et al. en una serie de 68 enfermos diagnosticados de PAI muestra que a 53 de ellos se les realizó resección pancreática como primera opción terapéutica y 15 de ellos no fueron intervenidos quirúrgicamente. En comparación con los pacientes no operados, el grupo de operados presentaba una menor proporción de aumento difuso del páncreas (80 vs. 8%, respectivamente) y menor número pretratamiento de análisis sérico de IgG4 (100 vs. 11%). La citología obtenida mediante punción fue erróneamente diagnóstica de adenocarcinoma en 12 pacientes, de los cuales 10 fueron intervenidos quirúrgicamente. Este estudio concluye que los factores que motivan la resección pancreática en pacientes con PAI por diagnóstico erróneo incluyen la dificultad en reconocer detalles propios de la enfermedad, como son manifestaciones radiológicas características, y los falsos positivos de la citología obtenida por ecoendoscopia (33). La limitante principal de esta prueba es el pequeño volumen de biopsia que se puede obtener, por lo que si no se lleva a cabo por endoscopistas experimentados, generalmente no es útil para el diagnóstico definitivo de PAI41,42.
Una necesidad imperiosa en la actualidad es el desarrollo de marcadores serológicos o pruebas de imagen que nos permitan identificar de manera fácil y precisa la enfermedad. A falta de ello, los esfuerzos se dirigen a desarrollar estrategias que pretenden facilitar el diagnóstico diferencial con el cáncer de páncreas mediante la combinación de elementos diagnósticos43–46. En este sentido también se han elaborado los criterios diagnósticos internacionales para consensuar un diagnóstico sistematizado en estos pacientes3. En la mayoría de casos, la evidencia de una masa hipointensa en la cabeza pancreática en un paciente con ictericia obstructiva no planteará ninguna duda respecto al diagnóstico de cáncer de páncreas. Sin embargo, es importante replantear del diagnóstico ante la existencia de hallazgos atípicos, como son por ejemplo, una evolución clínica atípica, la afectación de otros órganos, la existencia de halo periférico en la masa pancreática, la falta de dilatación ductal preestenótica o la elevación a niveles diagnósticos de la IgG4 sérica. Uno de los elementos diagnósticos reconocidos por el CIDC es la respuesta a corticoides. Esta es una herramienta que podemos considerar siempre y cuando hayamos excluido con un alto índice de seguridad la existencia de cáncer de páncreas; por ejemplo, en masas pancreáticas con una imagen radiológica atípica y citología negativa para células malignas. En casos como este se recomienda un periodo inicial de tratamiento de 2 semanas y volver a reevaluar la clínica y manifestaciones radiológicas del paciente47. Cabe mencionar también la evidencia descrita de casos de adenocarcinoma de páncreas en pacientes con PAI48,49 y de neoplasia papilar intraductal mucinosa50,51, lo que pone de manifiesto la necesidad de realizar seguimiento estrecho de estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Todos los autores participaron en la redacción, revisión crítica y aprobación de la versión final del artículo.

