







Familial hypercholesterolemia (FH) is the most common genetic disorder in childhood, but in most cases is not detected. High levels of low-density lipoprotein cholesterol are present since the child's birth and this fact will suppose silent development of early atherosclerosis. In cases of homozygous FH, the coronary disease will appear before 20s and in cases of heterozygous FH will occur in middle age. Despite published data, there is not agreement about how and when perform the screening. Familial history of early cardiovascular disease plus presence of hypercholesterolemia in parents is crucial for detection and diagnosis. Actually, it is topic of discussion that it is necessary to achieve therapeutic goals from an early age to improve prognosis. Lifestyle changes are the first line therapy. Statins are the lipid-lowering drugs of choice but the optimal age to start therapy it is still controversial. In this article, current recommendations of expert consensus guidelines about the management and new line therapies of child and adolescents are reviewed.
La hipercolesterolemia familiar (HF) es el trastorno genético más prevalente en edad pediátrica; sin embargo, en la inmensa mayoría de los casos pasa totalmente desapercibida. La elevación del colesterol ligado a las lipoproteínas de baja densidad presente desde el nacimiento comportará el desarrollo silente de arteriosclerosis de forma precoz. Este hecho podrá manifestarse en forma de enfermedad coronaria antes de los 20 años en la HF homocigota o en la edad media de la vida en la HF heterocigota. A pesar de las evidencias científicas, no existe un acuerdo común de cómo y cuándo se debe hacer el cribado, hecho que se pone de manifiesto al revisar las diferentes guías de consenso de expertos. La historia familiar de enfermedad cardiovascular prematura, junto con la presencia de hipercolesterolemia en uno de los progenitores, es crucial en la detección y el diagnóstico. Alcanzar los objetivos terapéuticos desde edades tempranas es un elemento clave en el pronóstico, aunque sigue siendo un tema de amplio debate. El primer eslabón del tratamiento siempre serán las recomendaciones de hábitos de vida cardiosaludables. En la actualidad, existe controversia sobre a qué edad se debe iniciar el tratamiento farmacológico, siendo las estatinas el fármaco de primera elección. En este artículo se revisan las recomendaciones actuales de las guías de consenso de expertos en el manejo del niño y adolescente con HF, así como las nuevas terapias emergentes.
Familial hypercholesterolaemia (FH) is a genetic autosomal dominant disorder, which means that there is a 50% chance of parent-to-child transmission. It is primarily caused by mutations in the gene that encodes the low-density lipoprotein receptor. At present, over 1700 different gene mutations have been described, representing over 90% of all FH cases. To a lesser extent, defects in the gene encoding Apolipoprotein B (ApoB) and the gene encoding proprotein convertase subtilisin/kexin type 9 (PCSK9) have been described, representing approximately 5% and 1% of cases, respectively. Clinically, these are expressed in the same way and only genetic testing can help us tell them apart. Nevertheless, in around 5–30% of current cases with the FH phenotype, it is not possible to identify the gene responsible for this disease.1
There are two types of FH: heterozygous (HeFH), which is the most common hereditary disorder with a prevalence of 1/250–500 individuals, and homozygous (HoFH), with a prevalence of 1/160,000–300,000 individuals, according to the most recent data published on the European population.2,3 Zamora et al. analysed the electronic records of 2,764,917 patients in Catalonia using the Big Data methodology. Using the cut-off level of low-density lipoprotein cholesterol (LDL-C), described previously in the Spanish population, 14,274 were found to have a FH-compatible phenotype. These data indicate a HeFH prevalence of 1/256 subjects in the Spanish population.4 Sánchez reported a HoFH prevalence of 1/495,000 subjects after analysing 16,744 genetic studies performed in Spain between 1996 and 2015.5
In Europe, it is estimated that the number of affected patients is around 4.5 million, of which 20–25% are children and adolescents. The fact that less than 10% are diagnosed is a problem of huge clinical significance. In countries with intensive genetic screening programmes, like the Netherlands, percentages of detection exceed 70%. In Spain, the rate of FH patients who have undergone testing and been diagnosed is around 6%.6 Individuals with FH are 100 times more likely to develop premature cardiovascular disease (PCVD) than unaffected individuals.7 It is estimated that 85% of men and 50% of women will present with some form of coronary event before the age of 65 if they do not receive adequate treatment.8 Children with HoFH have an increased risk of developing coronary heart disease before 20 years of age if they do not receive intensive treatment.2,9 As such, early diagnosis and treatment are important for prognosis and long-term outcome.
Patients affected by FH present high plasma concentrations of LDL-C, which may be detected from birth. Children with HeFH have LDL-C levels that are three-times higher than unaffected individuals10 and the condition may manifest as premature coronary heart disease in adulthood.2
In spite of all this, there is no clinical expression in childhood, thus necessitating the design and application of different strategies for its detection. The clinical criteria of the Dutch Lipid Clinic Network are not applicable to patients under the age of 18.11 In children, diagnostic suspicion should be established based on high LDL-C levels, family history of hypercholesterolaemia and/or PCVD.
The ideal age for detection is currently considered to be around 8–10 years, as this is the age of maximum discrimination. During adolescence, total cholesterol (TC) and LDL-C values decrease by around 10–20%, thus making this stage of life less sensitive for the performance of screening.12,13 Recently, Eissa et al. reported that lipid profiles vary considerably depending on the stage of pubertal development. They propose that this should be considered when screening is indicated.14 Pang et al. studied 1602 healthy adolescents between the ages of 14 and 17, defining FH according to LDL-C levels and family history of hypercholesterolaemia and/or PCVD. The prevalence of FH was 1/267 adolescents, with the authors underlining the absence of PCVD in parents, but the presence of PCVD in grandparents.15
Various studies have been published in which it has been observed that carotid artery intima-media thickness is higher in children with the FH phenotype than in normolipaemic children, as it is directly related to LDL-C levels. This significant difference in carotid artery intima-media thickness was observed in children from 7 years of age.2 The presence of coronary calcifications has also been observed in 25% of HeFH patients aged 11–23 years, and especially in the aortic artery of the majority of adolescents with HoFH.
Detecting FH in the paediatric population is still a major challenge, as the vast majority are undiagnosed, thus leading to a delay in the onset of treatment, both in terms of lifestyle changes and drugs, which may cause coronary risk to increase in this population during middle age.
The LDL-C cut-off level, the age at screening or the criteria for suspecting the condition in a parent are currently a source of dispute; this review thus describes different screening strategies for improving detection and diagnosis in children with FH. In turn, this review includes up-to-date recommendations on lifestyle changes and pharmacological treatment.
Children and adolescents with familial hypercholesterolaemiaWhy perform screening?Screening in childhood and adolescence is completely justified:
- -
Hypercholesterolaemia increases the risk of accelerated arteriosclerosis development and, as a result, PCVD.
- -
Screening may help to identify this high-risk population.
- -
At present, safe and effective pharmacological treatments are available that may slow down or even reverse arteriosclerosis, thereby decreasing cardiovascular (CV) risk in these children.
However, despite the evidence, there is currently no consensus. If we compare the strategies recommended on different continents, we can observe differences between them. Moreover, there are even discrepancies between the countries within Europe.7
Types of screening strategiesThere are different strategies—which are not mutually exclusive—for diagnosing new cases among the paediatric population.
Universal screening: consists of the routine measurement of TC levels in children of a certain age. This method has been proven to detect 90% of children with FH between the ages of 1–9, with a false positive ratio of less than1%.16 An example of this type of screening is the model applied in Slovenia at 5 years of age.17
Failing that, opportunistic screening is recommended, i.e. the inclusion of the TC assessment in any blood test prescribed by the paediatrician between 2 and 9 years of age.
Selective screening: consists of the measurement of TC levels in children with a family history of either PCVD or hypercholesterolaemia in one of the parents. This type of screening was recommended by the first paediatric expert panel, the National Cholesterol Education Program,17 by the American Heart Association18 and the American Academy of Pediatrics.19 However, it has become apparent that, on applying this screening method, between 30% and 60% of children with FH pass by undetected.
Direct cascade screening: if we know the genetic mutation responsible for the parent's FH, genetic testing is extended to all first-degree relatives, including children. This type of genetic screening boasts 100% sensitivity and specificity in family studies and is recommended as it is the technique that offers the best cost-effectiveness. A clear example of this type of screening is the model applied in the Netherlands from 1994 to the end of 2014.20,21 In the United Kingdom, guidelines recommend performing genetic testing in adult individuals and extending cascade screening to children over 10 years of age.7
In Spain, the SAFEHEART study detected a total of 1984 new relatives with FH from 768 probands. The authors highlight that, despite being a hereditary disorder, 25% of the detected relatives did not know that they were carriers of the disease and 20% did not receive treatment.22
Data from the study by Oliva et al. suggest that familial cascade screening with genetic testing and subsequent statin treatment is cost-effective.23
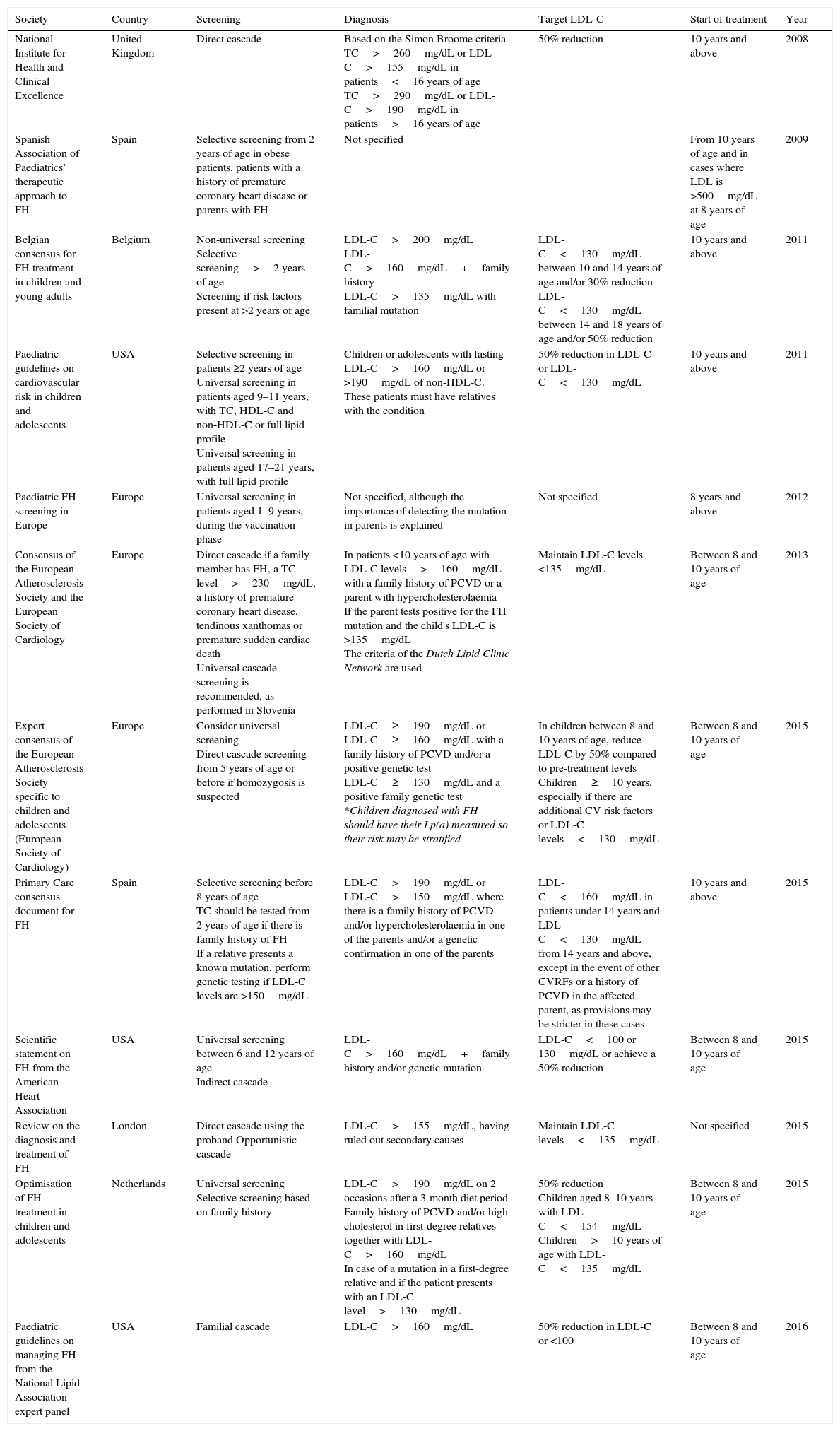
Reverse cascade screening: where testing is initiated in the parents after detecting hypercholesterolaemia in the child. If one parent presents a score≥6 in the clinical criteria of the Dutch Lipid Clinic Network, genetic testing will be requested. If the mutation is detected, genetic testing will be performed on the child. In contrast, if the result is negative, the child will not undergo testing, but he/she may be diagnosed with FH if LDL-C levels exceed the 95th percentile. Various guidelines recommend performing this type of screening, but it is underused in clinical practice. Table 1 depicts the up-to-date guidelines and the types of screening they recommend.
Summary of the main guidelines on familial hypercholesterolaemia in childhood and adolescence.
| Society | Country | Screening | Diagnosis | Target LDL-C | Start of treatment | Year |
|---|---|---|---|---|---|---|
| National Institute for Health and Clinical Excellence | United Kingdom | Direct cascade | Based on the Simon Broome criteria TC>260mg/dL or LDL-C>155mg/dL in patients<16 years of age TC>290mg/dL or LDL-C>190mg/dL in patients>16 years of age | 50% reduction | 10 years and above | 2008 |
| Spanish Association of Paediatrics’ therapeutic approach to FH | Spain | Selective screening from 2 years of age in obese patients, patients with a history of premature coronary heart disease or parents with FH | Not specified | From 10 years of age and in cases where LDL is >500mg/dL at 8 years of age | 2009 | |
| Belgian consensus for FH treatment in children and young adults | Belgium | Non-universal screening Selective screening>2 years of age Screening if risk factors present at >2 years of age | LDL-C>200mg/dL LDL-C>160mg/dL+family history LDL-C>135mg/dL with familial mutation | LDL-C<130mg/dL between 10 and 14 years of age and/or 30% reduction LDL-C<130mg/dL between 14 and 18 years of age and/or 50% reduction | 10 years and above | 2011 |
| Paediatric guidelines on cardiovascular risk in children and adolescents | USA | Selective screening in patients ≥2 years of age Universal screening in patients aged 9–11 years, with TC, HDL-C and non-HDL-C or full lipid profile Universal screening in patients aged 17–21 years, with full lipid profile | Children or adolescents with fasting LDL-C>160mg/dL or >190mg/dL of non-HDL-C. These patients must have relatives with the condition | 50% reduction in LDL-C or LDL-C<130mg/dL | 10 years and above | 2011 |
| Paediatric FH screening in Europe | Europe | Universal screening in patients aged 1–9 years, during the vaccination phase | Not specified, although the importance of detecting the mutation in parents is explained | Not specified | 8 years and above | 2012 |
| Consensus of the European Atherosclerosis Society and the European Society of Cardiology | Europe | Direct cascade if a family member has FH, a TC level>230mg/dL, a history of premature coronary heart disease, tendinous xanthomas or premature sudden cardiac death Universal cascade screening is recommended, as performed in Slovenia | In patients <10 years of age with LDL-C levels>160mg/dL with a family history of PCVD or a parent with hypercholesterolaemia If the parent tests positive for the FH mutation and the child's LDL-C is >135mg/dL The criteria of the Dutch Lipid Clinic Network are used | Maintain LDL-C levels <135mg/dL | Between 8 and 10 years of age | 2013 |
| Expert consensus of the European Atherosclerosis Society specific to children and adolescents (European Society of Cardiology) | Europe | Consider universal screening Direct cascade screening from 5 years of age or before if homozygosis is suspected | LDL-C≥190mg/dL or LDL-C≥160mg/dL with a family history of PCVD and/or a positive genetic test LDL-C≥130mg/dL and a positive family genetic test *Children diagnosed with FH should have their Lp(a) measured so their risk may be stratified | In children between 8 and 10 years of age, reduce LDL-C by 50% compared to pre-treatment levels Children≥10 years, especially if there are additional CV risk factors or LDL-C levels<130mg/dL | Between 8 and 10 years of age | 2015 |
| Primary Care consensus document for FH | Spain | Selective screening before 8 years of age TC should be tested from 2 years of age if there is family history of FH If a relative presents a known mutation, perform genetic testing if LDL-C levels are >150mg/dL | LDL-C>190mg/dL or LDL-C>150mg/dL where there is a family history of PCVD and/or hypercholesterolaemia in one of the parents and/or a genetic confirmation in one of the parents | LDL-C<160mg/dL in patients under 14 years and LDL-C<130mg/dL from 14 years and above, except in the event of other CVRFs or a history of PCVD in the affected parent, as provisions may be stricter in these cases | 10 years and above | 2015 |
| Scientific statement on FH from the American Heart Association | USA | Universal screening between 6 and 12 years of age Indirect cascade | LDL-C>160mg/dL+family history and/or genetic mutation | LDL-C<100 or 130mg/dL or achieve a 50% reduction | Between 8 and 10 years of age | 2015 |
| Review on the diagnosis and treatment of FH | London | Direct cascade using the proband Opportunistic cascade | LDL-C>155mg/dL, having ruled out secondary causes | Maintain LDL-C levels<135mg/dL | Not specified | 2015 |
| Optimisation of FH treatment in children and adolescents | Netherlands | Universal screening Selective screening based on family history | LDL-C>190mg/dL on 2 occasions after a 3-month diet period Family history of PCVD and/or high cholesterol in first-degree relatives together with LDL-C>160mg/dL In case of a mutation in a first-degree relative and if the patient presents with an LDL-C level>130mg/dL | 50% reduction Children aged 8–10 years with LDL-C<154mg/dL Children>10 years of age with LDL-C<135mg/dL | Between 8 and 10 years of age | 2015 |
| Paediatric guidelines on managing FH from the National Lipid Association expert panel | USA | Familial cascade | LDL-C>160mg/dL | 50% reduction in LDL-C or <100 | Between 8 and 10 years of age | 2016 |
Various FH reviews and consensuses have been published in recent years.
The Asociación Española de Pediatría [Spanish Association of Paediatrics] published guidelines for treating children with hypercholesterolaemia24 that can be summarised as follows:
- -
Universal screening is not recommended.
- -
Selective screening is recommended if there is a history of PCVD in first- and second-degree relatives and/or a TC value >240mg/dL in one parent.
- -
The ideal screening age is between 2 and 10 years.
The Sociedad Española de Arteriosclerosis [Spanish Arteriosclerosis Society] published an expert consensus document11:
- -
Universal screening in all children between 8 and 10 years of age.
- -
Direct cascade screening of first-degree relatives of patients with a genetic diagnosis of FH, irrespective of TC levels. If the genetic test comes back negative, LDL-C levels should be determined in all first-degree relatives.
- -
Reverse cascade screening of first-degree relatives of children with LDL-C levels >135mg/dL or established genetic testing.
- -
Selective screening of children with a family history of PCVD and/or hypercholesterolaemia.
The Fundación de Hipercolesterolemia Familiar [Familial Hypercholesterolaemia Foundation] published an expert consensus document25:
- -
Direct cascade screening from 2 years of age when one of the parents is diagnosed and, if possible, before 8 years of age.
- -
Suspect FH in children with LDL-C levels>190mg/dL or LDL-C>150mg/dL where there is a family history of PCVD and/or hypercholesterolaemia in one of the parents and/or a genetic confirmation in one of them.
The clinical diagnosis of FH in childhood can be difficult as a phenotypic overlap with polygenic hypercholesterolaemia can sometimes be observed. The child's diagnosis should preferably be genetic; however, this is not always possible. In this case, we will rely on the child's phenotypic expression and any family history of PCVD and/or hypercholesterolaemia indicating FH.
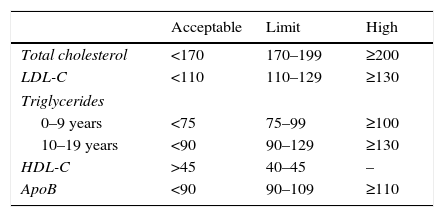
At present, in our field clinical suspicion will be based on family histories and TC levels. Table 2 shows the lipid profile considered to be within the normal and pathological range according to age.
Reference lipid profile in childhood and adolescence.
| Acceptable | Limit | High | |
|---|---|---|---|
| Total cholesterol | <170 | 170–199 | ≥200 |
| LDL-C | <110 | 110–129 | ≥130 |
| Triglycerides | |||
| 0–9 years | <75 | 75–99 | ≥100 |
| 10–19 years | <90 | 90–129 | ≥130 |
| HDL-C | >45 | 40–45 | – |
| ApoB | <90 | 90–109 | ≥110 |
Data expressed as mg/dL.
Source: Moráis-López et al.24
In light of TC levels≥200mg/dL, a second test is recommended within a maximum period of 3 months, requesting a full lipid profile. It would be advisable to also use the second blood draw to rule out secondary causes of hypercholesterolaemia in childhood (Table 3). If TC values≥200mg/dL and LDL-C levels≥130mg/dL are confirmed and secondary causes have been ruled out, we should recommend a period of dieting for no less than 6 months. After this period, if LDL-C levels≥130mg/dL persist in an additional test, we should suspect FH if accompanied by another condition (Table 4). If the child's test is the result of selective screening, his/her full lipid profile will be requested immediately.
Secondary causes of hypercholesterolaemia in childhood.
| Drugs: amiodarone, corticosteroids, anabolic steroids, cyclosporin, phenobarbital, progestogens, phenytoin, thiazides, etc. |
| Anorexia nervosa |
| Cholestasis: biliary cirrhosis, biliary atresia |
| Growth hormone deficiency |
| Endocrine disorders: hypothyroidism, hypopituitarism |
| Kidney diseases: nephrotic syndrome |
| Idiopathic hypercalcaemia |
| Acute intermittent porphyria |
| Deposition diseases: glycogenosis, Tay-Sachs, Gaucher, Niemann-Pick |
| Dietary factors |
Suspicion criteria for familial hypercholesterolaemia in childhood and adolescence.
| Child with LDL-C ≥130mg/dL and one of the following: |
| History of PCVD in first-degree relatives <55 years of age and second-degree relatives <50 years |
| Parent with total cholesterol levels >300mg/dL or receiving lipid-lowering treatment |
| Lack of information on the parents |
| One parent affected by FH with a clinical or genetic diagnosis |
If LDL-C levels are ≥190mg/dL, obtained over 2 consecutive tests with a 3-month gap, the probability of finding a causative mutation for FH is very high. If LDL-C levels are ≥160mg/dL after a period of dieting and there is a history of PCVD in first-degree relatives (men<55 years, women<60 years) or second-degree relatives (men<45 years, women<50 years) and/or hypercholesterolaemia (c-LDL≥190mg/dL) or one of the parents is receiving a lipid-lowering pharmacological treatment, there is a high probability of them being a carrier of a causative mutation for FH. If the causative mutation has been detected in one of the parents and the child has LDL-C levels≥130mg/dL, he/she is also very likely to carry this mutation. If the parent has died due to PCVD and the child presents with LDL-C levels≥130mg/dL, genetic testing should be attempted.
Genetic testing is recommended. However, at present this is not possible in the entire Spanish population and there are large differences between autonomous communities, with very different circumstances and realities. A clear example of this is that, while in the Canary Islands and Murcia, genetic testing is not subsidised, in Castile and León, there is no form of restriction.26
LDL-C levels in children with HeFH range between 190 and 500mg/dL while, in those with HoFH, this figure ranges between 500 and 1000mg/dL, together with the presence of tuberous xanthomas and arcus senilis before the age of 10. However, an overlap of LDL-C levels of between 300 and 500mg/dL can be seen in both forms.27
Recently, a computer program has been designed that calculates the probability of detecting a mutation in a patient with suspected FH and which aims to increase detection in young people. This tool is based on the data collected over a 20-year period in the Dutch FH detection programme (http://vasculaironderzoekamc.nl/fh-calculator/).28
Therapeutic objectivesThere is no evidence that indicates therapeutic objectives in children with FH. Table 1 summarises the different therapeutic objectives in relation to the various guidelines published in recent years.
Therapeutic recommendationsIt is important to initiate therapy early in children with FH.29
Lifestyle changesA balanced and healthy diet is key to FH treatment and in order to prevent arteriosclerosis. It has been observed that diet may reduce LDL-C levels by 10–15%, although this can vary significantly according to the type of patient and type of mutation.24,25 It is worth noting that, in these children, dietetic recommendations alone are not going to be sufficient to achieve the therapeutic objectives; nonetheless, maintaining an appropriate weight remains important in order to avoid adding further CV risk factors.30
These recommendations should be indicated from 2 years of age under the supervision of a qualified dietician/nutritionist who helps to reinforce nutrition therapy in the family setting in order to achieve better treatment adherence.31
FatFat consumption should be limited to <30% of the total calorie intake. Until recently, many of the guidelines published specified the need to lower all types of fat and to place special emphasis on the importance of limiting cholesterol intake (<200–300mg/day). However, more stress is now being placed on quality over quantity.
The intake of saturated fatty acids should be <10%; however, some guidelines are stricter and state it is best to stay below 7%. Reducing saturated fat and cholesterol in children's diets has not been shown to alter their nutritional status, growth or pubertal development.32 Their monounsaturated fat intake should be kept at around 10%, principally in the form of oleic acid. The trans fats found in processed foods should be avoided.
Different clinical trials have shown that the daily consumption of plant stanols and sterols at doses of 1.5–3g/day in children and adolescents with HeFH can be beneficial in reducing LDL-C levels by approximately 9–19%.33 Recently, the European Atherosclerosis Society published a consensus panel which recommends consumption of plant stanols and sterols in FH patients from 6 years of age, as long as their fruit and vegetable intake is sufficient in order to avoid a fat-soluble vitamin deficiency.34
None of the following types of supplement is recommended: linoleic acid, omega 3, rapeseed oil,35 soy protein, garlic extracts36 or cereals containing psyllium extract.37
CarbohydratesCarbohydrates include fibres, starches and sugars. They can also be categorised into two types: simple and complex. Complex carbohydrates should be promoted as they have a lower percentage of calories and a high fibre content (whole grains, pasta, rice, bread, potatoes, legumes, fruit and vegetables).
The latest European guidelines on cardiovascular disease prevention advise a fibre intake of 30–45g/day. The consumption of water-soluble fibre in the form of fortified cereals can be added to diets that are low in fat or saturated fat, and the recommended daily dose is 6g/day for children aged 2–12 and 12g/day for those over 12.
The intake of simple sugars should also be reduced, along with the sugar content in soft drinks.38,39
ProteinsA protein intake of around 15% should be advised. Preference should be given to the consumption of white meat and fish.
SaltMany of the clinical guidelines recommend a maximum intake of 5g/day (2g of sodium). In children under 10 years of age, less than 3–4g/day is advisable; however, the average consumption among this age group is 8.1g/day.40 Around 75% comes from processed foods.
AlcoholAmong the school population aged between 14 and 18 years, the most widely consumed drink during the week is beer; at the weekends, spirits and mixers are most common.
It is important to teach children not to consume alcohol.
Physical exerciseMaintaining an ideal weight and encouraging physical exercise are key factors among the set of measures to be followed.
Ideally, children should exercise for over an hour every day, and should spend less than 2h performing sedentary activities such as playing on consoles or computers and/or watching TV.4
It has been observed that physical exercise patterns established during childhood remain lifelong habits and are associated with increased high-density lipoprotein cholesterol and reduced LDL-C.41,42
SmokingThe European CV prevention guidelines propose different strategies for promoting smoking cessation.13 Tobacco consumption has decreased by 60% in the last decade according to the report issued by the Spanish Ministry of Health; nevertheless, 38.4% of Spanish teenagers admit to having smoked at least once and 8.9% do so on a daily basis.
Smoking is another CV risk factor and this risk is even higher in patients with FH.43
Pharmacological treatmentEfficacy and tolerance of statinsStatins are the drugs of choice in the pharmacological management of children with FH. They will always be preceded by a period of dietary treatment. Pharmacological treatment should start before puberty as this improves control and parental adherence, with the child's acceptance also being greater.
Statins act by competitively inhibiting the 3-HMG-CoA reductase enzyme, leading to a decrease in intrahepatic cholesterol synthesis and an increase in LDL receptor synthesis.
Various expert consensus guidelines recommend starting statin treatment at 8–10 years of age in children with HeFH, and at the time of diagnosis in children with HoFH, before 5 years of age and no later than 8.27
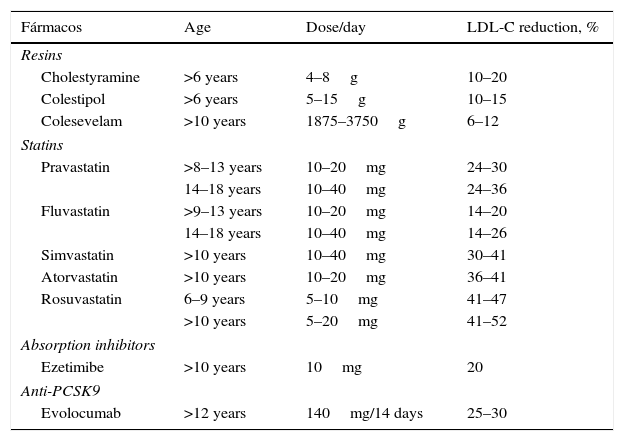
The statins shown below are currently approved by the US Food and Drug Administration and the European Medicines Agency (Table 5). Pitavastatin has not been approved in children, although a recently-published study demonstrates its safety in patients aged 6–17 years.44 Various studies have shown the short-term safety and efficacy of statins. A follow-up study was recently published on a cohort of 214 children with FH treated with pravastatin for 10 months, demonstrating that the long-term safety does not differ from that reported in adult patients.45
Pharmacological treatment of children and adolescents with familial hypercholesterolaemia.
| Fármacos | Age | Dose/day | LDL-C reduction, % |
|---|---|---|---|
| Resins | |||
| Cholestyramine | >6 years | 4–8g | 10–20 |
| Colestipol | >6 years | 5–15g | 10–15 |
| Colesevelam | >10 years | 1875–3750g | 6–12 |
| Statins | |||
| Pravastatin | >8–13 years | 10–20mg | 24–30 |
| 14–18 years | 10–40mg | 24–36 | |
| Fluvastatin | >9–13 years | 10–20mg | 14–20 |
| 14–18 years | 10–40mg | 14–26 | |
| Simvastatin | >10 years | 10–40mg | 30–41 |
| Atorvastatin | >10 years | 10–20mg | 36–41 |
| Rosuvastatin | 6–9 years | 5–10mg | 41–47 |
| >10 years | 5–20mg | 41–52 | |
| Absorption inhibitors | |||
| Ezetimibe | >10 years | 10mg | 20 |
| Anti-PCSK9 | |||
| Evolocumab | >12 years | 140mg/14 days | 25–30 |
Treatment should be started if LDL-C levels are ≥190mg/dL or ≥160mg/dL if accompanied by one of the following conditions: history of PCVD in a first-degree relative; other diseases suffered by the child that involve elevated CV risk (diabetes, metabolic syndrome, hypertension, lupus erythematosus, organ transplant and Kawasaki disease); or in the event of high CV risk factors (obesity, smoking, increased Lp(a) and homocysteinaemia).13
When statin treatment is initiated, the lowest recommended dose should be chosen, which can then be adjusted according to response and tolerance. In any case, combination therapy with other drugs (ezetimibe or resins) may be necessary in order to achieve the therapeutic objectives. Table 6 shows the parameters to be monitored. Although side effects are uncommon, the following have been reported: cramps, muscle pain, gastrointestinal effects and elevated transaminases. No growth hormone or sex hormone abnormalities has been reported.46
Recommendations before and after statin use in children.
| Prior to starting the treatment, determine the patient's liver transaminases, CPK, creatinine and glucose |
| After starting the treatment, monitor the patient's lipid profile and liver transaminases |
| Monitor CPK if the patient reports myalgia |
| Monitor glucose in patients receiving high statin doses and who are obese |
| Growth and pubertal development check |
| Yearly check once the therapeutic objective has been met |
If transaminases are observed to be three times the upper limit of normal, the dose should be reduced or withdrawn and an additional follow-up check should be arranged. If the child undertakes intensive exercise, we recommend stopping exercise at least three days before blood draws in order to avoid elevated CPK levels secondary to exercise.
In children that require pharmacological treatment due to other concomitant diseases, it is important to recognise that cytochrome P450 uses the drug to be metabolised. The decision regarding the type of statin to be chosen for initiating treatment will depend on this fact. If the child presents with kidney failure, the statin of choice shall be atorvastatin, as its excretion by the kidneys is minimal. The paediatrician should be aware of potential drug interactions with statins and must avoid prescribing routine drugs like macrolides.
Girls of childbearing age should be advised on different methods of contraception and must be informed that accidental pregnancies are to be avoided. In case of accidental pregnancy, pharmacological treatment must be immediately suspended.
Efficacy and tolerance of ezetimibeEzetimibe is a drug that acts by selectively inhibiting cholesterol and plant sterol absorption in the small intestine, without affecting the absorption of fat-soluble vitamins or other substances.
In general, it is a second-line, lipid-lowering drug that is usually administered in combination with a statin. It is indicated in monotherapy in the event of statin intolerance. Experience of the drug in children is somewhat limited; the studies that have been conducted have only evaluated its short-and medium-term safety and efficacy and, as such, more studies are needed to assess its long-term profile47,48 (Table 5).
Efficacy and tolerance of resinsIon-exchange resins act by inhibiting bile acid absorption in the intestine.49
There are a number of resins that are currently available on the market (Table 5). However, gastrointestinal intolerance and difficult ingestion can lead to discontinuation of treatment. In an attempt to reduce this problem, dissolving the powder or granules in fruit juice and taking them before meals is recommended. It must be remembered that the drug cannot be administered together with other treatments (1h before or 4h after). Prolonged treatments may alter the absorption of fat-soluble vitamins (A, D, E and K) and folic acid, so supplements may be required. The most common side effect is constipation, so a fibre-rich diet will be recommended.
Colesevelam is the newest resin to be placed on the market. Its safety and efficacy have been assessed in children with HeFH between the ages of 10 and 17 years. Due to its presentation, it has a seemingly greater tolerance than the other resins.50,51
Efficacy and tolerability of PCSK9 inhibitorsPCSK9 is a protein segregated by the hepatocytes, which intervenes in regulating cholesterol metabolism.52
PCSK9 inhibition reduces the number of receptors that are due to be degraded and thus increases their density on the cell surface, with the subsequent reduction of plasma cholesterol.
Anti-PCSK9 antibodies are a new group of drugs that have already proven their excellent short-term safety and efficacy. At present, evolocumab and alirocumab have been approved in Spain and are indicated for the FH population in which therapeutic objectives are not met at the maximum tolerated dose of lipid-lowering drugs.53,54
Children with HoFH have also been included in clinical trials with evolocumab. Additional LDL-C reductions have been observed in children carrying defective genes; however, no effect was observed on carriers of receptor-negative mutations.55,56 PCSK9 inhibitors will undoubtedly be a therapeutic option for a very specific group of children with HoFH, which could even lead to fewer LDL apheresis sessions for those administered this treatment (Table 5).
Future therapeutic innovationsLomitapideLomitapide is an oral inhibitor of the microsomal triglyceride transfer protein present in the endoplasmic reticulum of hepatocytes and enterocytes. It has been approved by the US Food and Drug Administration and the European Medicines Agency for the treatment of HoFH. Compared to the standard therapy, one study found lomitapide to reduce the LDL-C concentration by up to a further 50% after 12 months of follow-up.57 Side effects are very common and primarily of a gastrointestinal nature. Elevated transaminases and the onset of hepatic steatosis have been reported, which must therefore be monitored throughout treatment.
In Spain, the first clinical experience with lomitapide has been published.58 There are still no data available on children, although it is approved on a “compassionate use” basis.
MipomersenMipomersen is an antisense oligonucleotide that inhibits the mRNA transcription of ApoB. The reduction in ApoB synthesis gives rise to a decrease in intrahepatic VLDL and, consequently, LDL-C. A long-term study was recently published where treatment with this drug was associated with a reduction in CV events in patients with FH.59
It has not been approved by the European Medicines Agency due to the onset of numerous adverse effects, principally comprising local reactions at the injection site and elevated transaminases.60
Homozygous familial hypercholesterolaemiaHoFH is the form with the most severe expression. If they do not receive treatment, most children will go on to develop arteriosclerosis before 20 years of age and their life expectancy will not exceed 30 years.27
Starting treatment as early as possible is recommended in an attempt to delay the onset of CV disease. The therapeutic objective is the same in children with HeFH; however, in the event of established CV disease, the objective should be LDL-C levels<70mg/dL. These objectives are very hard to achieve. An oral lipid-lowering treatment should be administered as early as possible from 2 years of age, combining statins with ezetimibe or resins. Despite this, the LDL-C reductions observed will be very mild.
Most children will need to receive LDL apheresis. This is a method of extracorporeal ApoB-lipoprotein elimination, obtaining further LDL-C reductions of 55–70%. This reduction is temporary, so weekly or fortnightly sessions should be performed. It is recommended that apheresis be initiated in children below 5 years of age and never later than 8. LDL apheresis is well tolerated, with adverse effects reported in less than 5% of the procedures. One problem in younger children is venous access. On occasions, fitting a central access device will be necessary, with complications in the form of infections and thrombosis having been reported.
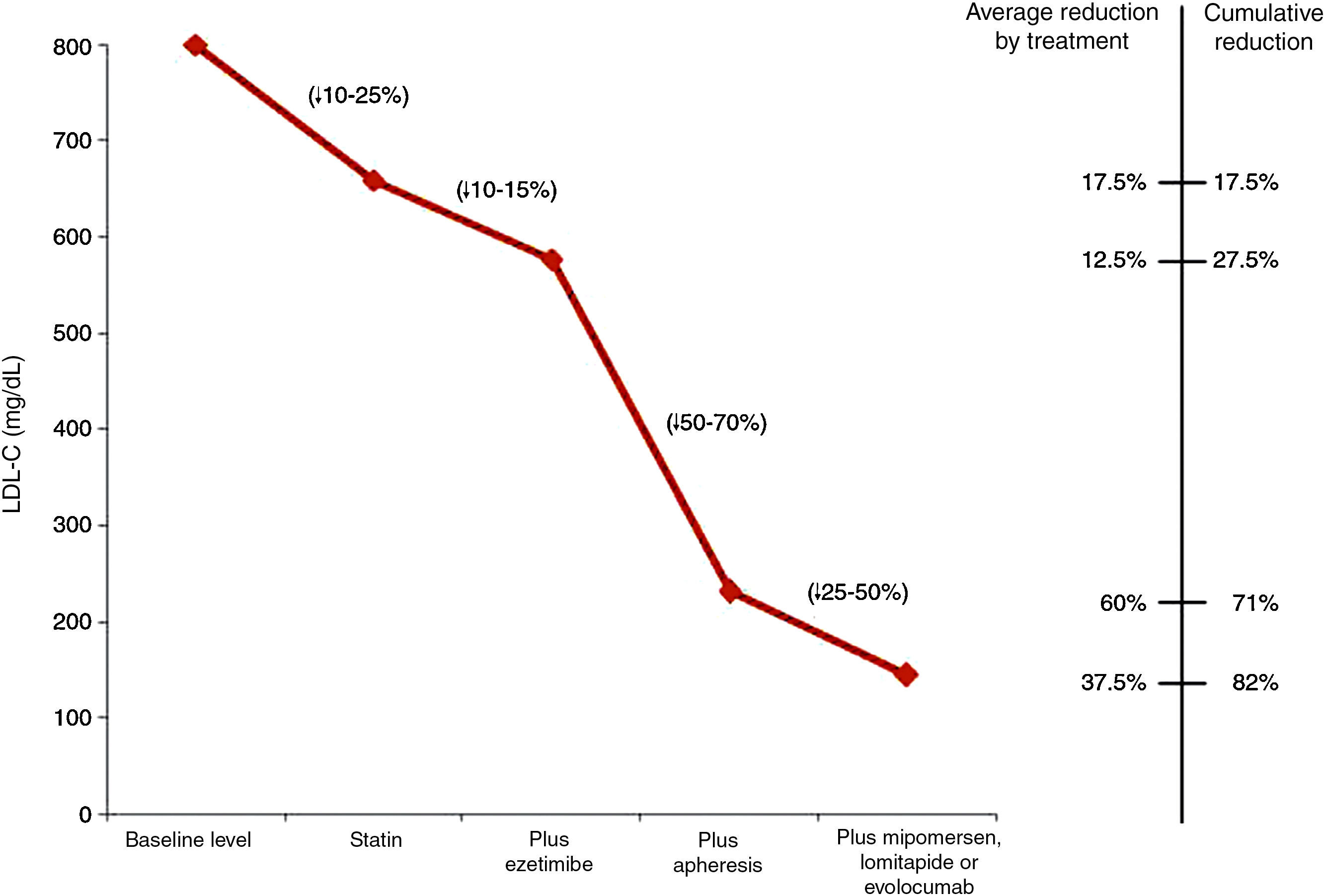
Fig. 1 shows the LDL-C reductions observed according to the treatment administered to children with HoFH.27

LDL cholesterol reduction according to the treatment administered to children with homozygous familial hypercholesterolaemia.27
Children with HoFH should be transferred to specialist units for treatment.
ConclusionsFH is a hereditary genetic disorder with a high CV risk. In order to prevent morbidity and mortality in later life, detection should be initiated in childhood. We believe that the paediatrician plays a vital role and universal screening in children under the age of 10 years would be advisable, although this practice is not accepted across the board at the present time. As an alternative, we recommend opportunistic screening. Direct cascade and selective screening should be performed in routine clinical practice. Reverse cascade screening has also proven to be an effective tool, and TC should be assessed with all blood draws performed. Healthy lifestyle habits should be encouraged in children from a young age and pharmacological treatment initiated whenever indicated. The performance of genetic testing is important in order to stratify risk, but also as a method of family adherence.
Children with FH should be transferred to specialist referral units with the capacity to perform genetic testing and experience in managing such cases. Children with HoFH will require intensive pharmacological treatment at maximum doses alongside LDL apheresis.
Ethical responsibilitiesProtection of people and animalsThe authors declare that no experiments were conducted on human beings or animals for this research.
Data confidentialityThe authors declare that no patient data is contained in this article.
Right to privacy and informed consentThe authors declare that no patient data is contained in this article.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Plana N, Rodríguez-Borjabad C, Ibarretxe D, Masana L. Hipercolesterolemia familiar en la infancia y la adolescencia: una realidad oculta. Clin Invest Arterioscler. 2017;29:129–140.