




The sellar and parasellar region is a complex anatomical area in which several diseases may develop. The pituitary gland may be affected by a wide range of conditions having similar clinical characteristics. Diagnosis of these lesions requires a multidisciplinary approach including, in addition to clinical, laboratory, imaging, and surgical findings, histological diagnosis of pituitary adenomas to guide therapeutic management. As the result of development in recent years of new immunohistochemical techniques, histopathological classification has become more complex and wide, and not only continues to be the gold standard in diagnosis, but also has prognostic implications. The aim of this review is to provide a clear and simple update of the main concepts of histological diagnosis of the most common pituitary conditions, especially for professionals in direct contact with such diseases.
La región selar y paraselar es una área anatómica compleja en la que se pueden desarrollar una serie de enfermedades. La glándula hipofisaria puede verse afectada por una amplia gama de trastornos, que cursan con características clínicas similares. El diagnóstico de estas lesiones implica un enfoque multidisciplinar y, junto con la exploración clínica, analítica, radiológica y quirúrgica, el estudio histológico de los adenomas hipofisarios determina la conducta que tomará el médico especialista ante el paciente. Con la aparición, en los últimos años, de nuevas técnicas inmunohistoquímicas, la clasificación histopatológica se ha vuelto más compleja y amplia, ya que además de ser el gold standard del diagnóstico, tiene implicaciones pronósticas. El objetivo de esta revisión es actualizar conceptos del diagnóstico histológico de la patología hipofisaria más frecuente, de manera clara y fácil, especialmente para aquellos profesionales en contacto directo con este tipo de patología.
The approach to the pathology of the pituitary gland and the sellar region is complex, because this area may be affected by many tumors and pseudotumoral lesions, and knowledge of multiple pathological conditions is therefore required. Tumors of the pituitary gland and sellar region account for approximately 15% of all brain tumors.1 The vast majority of them are pituitary adenomas (PAs) (85%), followed by craniopharyngiomas (3%), Rathke cleft cysts (2%), meningiomas (1%), and metastases (0.5%). All other tumors are very rare lesions2 that mimic PAs in neuroimaging studies, so that the final diagnosis should be made by the pathologist.
The development and widespread use of neuroradiological, computerized tomography, and magnetic resonance imaging studies has resulted in the increasingly frequent diagnosis of clinically silent pituitary lesions.3–5 Magnetic resonance imaging (MRI) is currently considered the preferred modality for the diagnosis of pituitary lesions because of its capacity to examine multiple planes and because of the possibility of differentiating soft tissues based on contrast uptake. A focal hypointensity inside the pituitary gland is considered abnormal and suggests an adenoma.
Many pseudotumoral and tumoral types of lesions may affect the pituitary gland and the sellar region (developmental abnormalities, cysts, inflammatory, infectious, metabolic, and neoplastic diseases, and vascular disorders), reflecting the complex anatomy of this area. This review will focus on the histological diagnosis of the most common and relevant pituitary conditions.
Tumors of the adenohypophysisGeneral characteristics of pituitary adenomasIncidental PAs may be found in approximately 10% of autopsies.6–8 In a recent review of autopsy and MRI studies, the estimated overall prevalence of PA was 16.7%.9 Primary tumors of the neurohypophysis are comparatively more uncommon, and usually similar to primary tumors of the central nervous system. However, the neurohypophysis is a common site for metastases.10
PAs are benign epithelial tumors derived from intrinsic cells of adenohypophysis. They occur in both sexes, predominantly between the third and sixth decades of life,11 but may affect any age group.1,12 Pediatric PAs are extremely rare, but when they do occur, they are usually ACTH-secreting adenomas.13 PAs are not homogeneous; each subtype has its own clinical presentation, trend to invasion, hormone secretion pattern, histopathological characteristics, and treatment. The mechanisms involved in tumor genesis and progression are not yet well known.
Clinically, PAs are classified as functioning and non-functioning depending on whether or not there is a specific endocrine syndrome. Approximately one third of PAs are not associated with any clinical or biochemical evidence of excess hormones14; they are clinically non-functioning adenomas, usually presenting with signs and symptoms related to the local mass effect such as headache, neurological deficits of the cranial nerves (including visual field changes), and hyperprolactinemia. Hyperprolactinemia is due to pituitary stalk compression (the so-called “stalk effect”), that prevents dopamine arrival to the adenohypophysis (and should not be misinterpreted by the pathologist as a prolactin-secreting adenoma).
Based on size and anatomical characteristics, adenomas are classified as microadenomas (<1cm in diameter), macroadenomas (>1cm to <4cm), and giant adenomas (>4cm). Radiographically, several classifications have been proposed to assess adenoma extension and local invasiveness. The Hardy and Knosp classifications are among those most commonly used.15,16
PAs are also classified histopatologically based on the hormonal content of tumor cells as shown by immunohistochemistry (IHC), which provides highly relevant information for clinical practice.17 In this article, the classification of pituitary gland tumors published in 2004 by the World Health Organization (WHO) will be followed.18
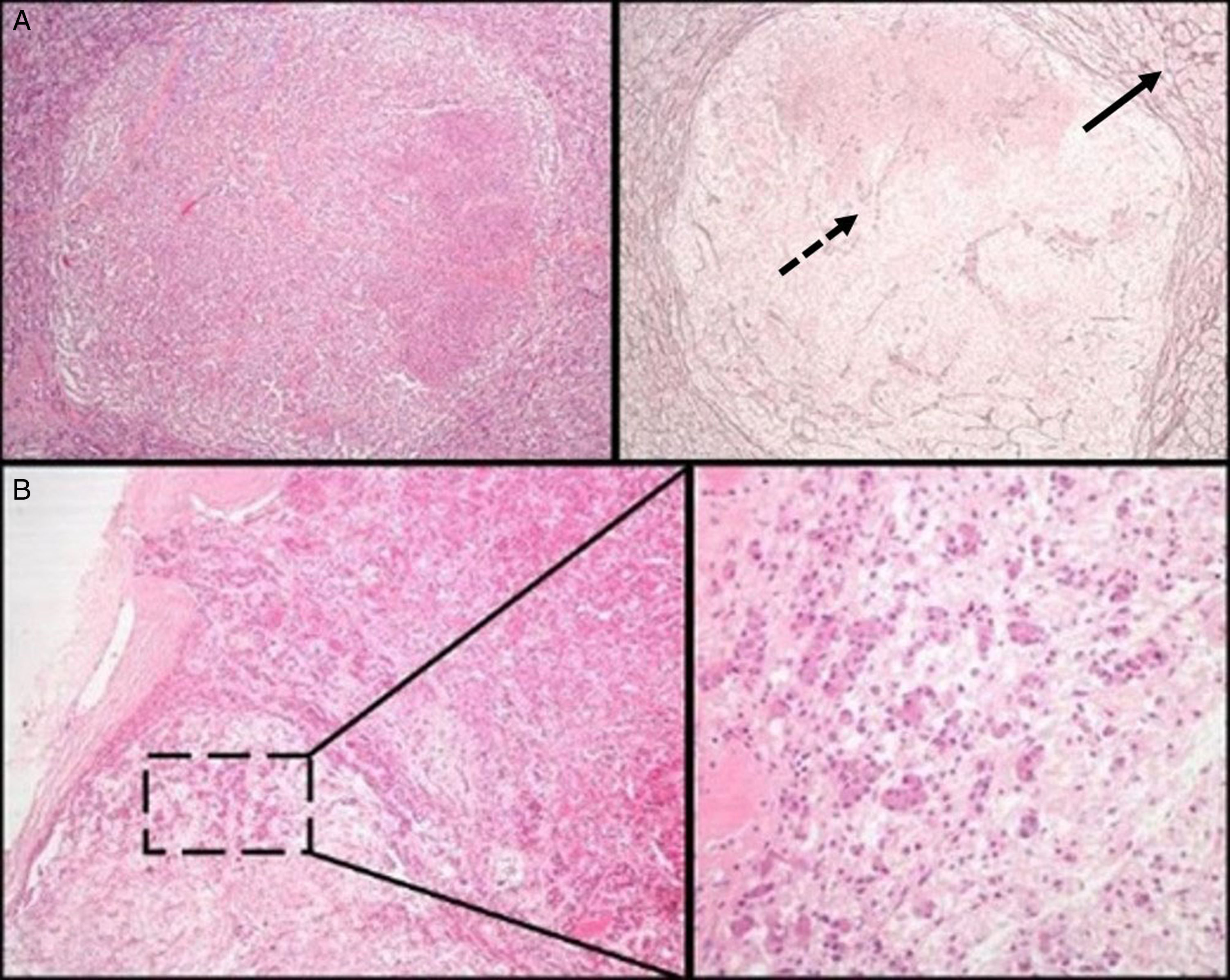
Initial pathological assessment of pituitary lesionsThe first decision to be taken when faced with a surgical specimen of the pituitary gland concerns whether the tissue submitted for analysis is a normal pituitary gland or a PA. For this, the most helpful histochemical stain after hematoxylin–eosin (HE) is the reticulin technique, which helps differentiate the preserved acinar pattern of normal adenohypophysis from the disruption of the reticulin network seen in PAs19 (Fig. 1A). HE and other special histochemical procedures, such as the periodic acid-Schiff (PAS)-orange G technique (now considered obsolete and widely replaced by IHC), help visualize the variety of cell types with different cytoplasm staining characteristics (acidophilic, basophilic, or chromophobic) present in normal adenohypophysis. In IHC, both normal adenohypophysis and PAs are immunoreactive to synaptophysin (a marker of neuroendocrine tumors); positivity for specific pituitary hormones demonstrates the great cellular variety seen in fragments of normal anterior pituitary gland (in contrast to what occurs in most PAs). Small fragments of normal neurohypophysis may sometimes be found, especially if the neurosurgeon has resected a Rathke cleft cyst. The best IHC technique to confirm the presence of posterior pituitary gland is neurofilament labeling, which permits us to differentiate it from other lesions, such as pituicytoma. The excision of small fragments of neurohypophysis usually has no permanent clinical consequences, but causes a transient diabetes insipidus that normally resolves in a few days. A variant of normality in the posterior pituitary gland that should not be confused with tumor infiltration, the so-called “basophil invasion” characteristic of aging, consisting of normal pituicytes from the anterior pituitary gland immunoreactive to ACTH that extend to neurohypophysis, should be kept in mind8 (Fig. 1B).
 Normal pituitary versus pituitary adenoma. Note the peripheral acinar pattern of the normal anterior pituitary gland (continuous arrow), in contrast to the reticulin network disruption common in an adenoma (dotted arrow) (histochemical technique: HE–left–and Gomori-reticulin–right–, 40×). (B) The normal pituitary gland shows a physiological “basophilic invasion” with aging. Streaks of basophilic endocrine cells are seen extending from the interphase of the anterior lobe to the neurohypophysis (HE 40×; HE 200×).")
(A) Normal pituitary versus pituitary adenoma. Note the peripheral acinar pattern of the normal anterior pituitary gland (continuous arrow), in contrast to the reticulin network disruption common in an adenoma (dotted arrow) (histochemical technique: HE–left–and Gomori-reticulin–right–, 40×). (B) The normal pituitary gland shows a physiological “basophilic invasion” with aging. Streaks of basophilic endocrine cells are seen extending from the interphase of the anterior lobe to the neurohypophysis (HE 40×; HE 200×).
The second decision to be taken will be if the lesion is a PA or not. The great majority of these tumors show a diffuse growth pattern. However, there may be occasional variations in their architecture (sinusoidal, macronodular, or scalloped pattern) that are not related to prognosis, but may confound diagnosis. Other potential characteristics include cells with clear cytoplasm, cysts of varying size, clefts caused by cholesterol crystals, xanthomatous macrophages, and even adaptive processes such as bone metaplasia (which should be distinguished from bone invasion of the floor of the sella turcica by the adenoma, which usually causes no osteoblast reaction and in which bone trabeculae become thinner).19
As regards the specific adenohypophyseal hormones needed for PA subtyping, we recommend antibodies to PRL, GH, ACTH, FSH, LH, and TSH as a minimum IHC panel. Prognostic markers, specifically the cell proliferation marker Ki-67 and the marker of the p53 tumor suppressor gene, are added for the differential diagnosis of typical and atypical PAs. As it is sometimes difficult to differentiate apoptotic nuclei from mitosis, the use of the phosphohistone H3 (PHH3) antibody is also recommended; once histone H3 (a protein in the nucleus of histone, the main protein constituent of chromatin) is not phosphorylated during apoptosis,20 it may serve to separate mitotic figures from apoptotic bodies and karyorrectic debris.
The use in IHC of vimentin, glial fibrillary acidic protein, or protein S-100 is of no value for PA diagnosis and subtyping, and is not recommended in the initial basic IHC. They may however be used when tumor characteristics under the light microscope suggest a spindle cell lesion in the sellar region.
Prolactin-secreting adenomasProlactin-secreting adenomas, also called prolactinomas, account for approximately 80% of functioning adenomas and for approximately 40%–50% of all Pas.21,22 They are the most common type of PA.23 However, the prevalence of prolactinoma in surgical series tends to be low because of their good response to medical treatment (most patients with prolactinoma are administered first-line treatment with dopamine agonists). Diagnosis is confirmed by sustained hyperprolactinemia and neuroradiological evidence of a pituitary tumor, other causes of hyperprolactinemia having been ruled out. Histologically, IHC shows reactivity for PRL, with a characteristic dot-like perinuclear labeling pattern known as the Golgi pattern. Dopamine agonists are drugs acting directly on tumor cells inducing atrophy and consequent tumor shrinkage. In these cases, histological analysis may reveal smaller tumor cells with cytoplasm reduction and hyperchromatic nuclei, in addition to different degrees of perivascular and interstitial tumor fibrosis. Prolactin identification in adenomas by IHC supports postoperative treatment with dopamine agonists if residual tumor or hypersecretion persists. In ultrastructural analysis, prolactinomas may be defined as either densely or sparsely granulated, although the clinical significance of this distinction is questionable.24
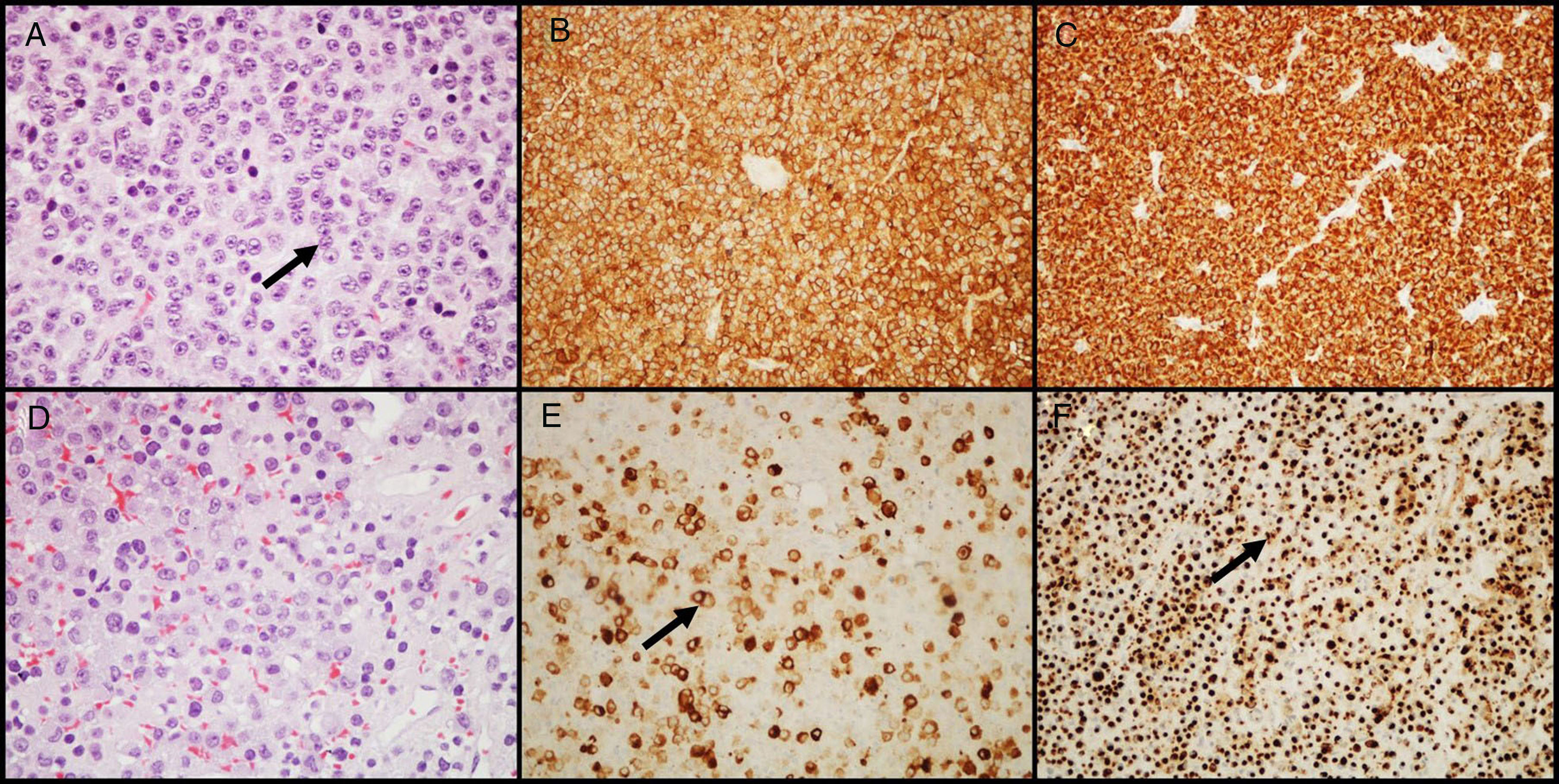
GH-secreting adenomasGH-secreting adenomas account for approximately 20% of PAs. Patients have signs and symptoms of gigantism or acromegaly, as well as high serum levels of GH and IGF-I.11 Prolactin co-secretion by the tumor is found in approximately 30%–50% of patients and results in signs and symptoms of hyperprolactinemia. Histologically, the amount of secretory granules present in cell cytoplasm characterizes two types of adenomas: densely granulated adenomas (characterized by an eosinophilic granular cytoplasm of tumor cells) and sparsely granulated adenomas (consisting of smaller tumor cells with chromophobic cytoplasms and eccentric nuclei). In the first group, IHC shows intense and diffuse staining for GH and the transcription factor Pit-1. In the second group, labeling for GH is heterogeneous and less intense, and cytoplasm may show paranuclear eosinophilic structures called “fibrous bodies”25 (an accumulation of intermediate filaments and endoplasmic reticulum), better visualized with IHC for cytokeratins 8/18 (Fig. 2). E-cadherin is another antibody that may be helpful in differentiating between them, because there is a loss of expression in sparsely granulated GH-secreting adenomas, but not in those densely granulated.26 The distinction between these two subtypes of adenoma is important because they show different clinical behavior (sparsely granulated adenomas have a more aggressive biological behavior and respond less to treatment with somatostatin receptor ligands).27,28 A large number of GH-secreting adenomas may show secondary immunoreactivity for other pituitary hormones (PRL, FSH, LH or β-TSH).27,29 In adenomas treated with somatostatin receptor ligands, mainly octreotide, the most common changes are various degrees of perivascular and interstitial fibrosis.
 Densely granulated GH-secreting adenomas show large cells with eosinophilic granular cytoplasm and a central nucleus with prominent nucleoli (arrow); (B) the tumor shows intense and diffuse immunolabeling for GH; (C) immunolabeling with cytokeratin shows diffuse cytoplasmic reactivity. (D) Sparsely granulated GH-secreting adenomas are characteristically more chromophobic than densely granulated adenomas; (E) GH labeling is heterogeneous and less prominent (arrow); (F) immunohistochemistry with cytokeratin highlights fibrous bodies (arrow). (A and D–HE 400×; B and E–GH 200×; C and F–cytokeratins 8/18 200×).")
GH-secreting adenomas. (A) Densely granulated GH-secreting adenomas show large cells with eosinophilic granular cytoplasm and a central nucleus with prominent nucleoli (arrow); (B) the tumor shows intense and diffuse immunolabeling for GH; (C) immunolabeling with cytokeratin shows diffuse cytoplasmic reactivity. (D) Sparsely granulated GH-secreting adenomas are characteristically more chromophobic than densely granulated adenomas; (E) GH labeling is heterogeneous and less prominent (arrow); (F) immunohistochemistry with cytokeratin highlights fibrous bodies (arrow). (A and D–HE 400×; B and E–GH 200×; C and F–cytokeratins 8/18 200×).
Mixed GH- and PRL-secreting adenomas account for approximately 8% of Pas.2 Patients with these tumors show signs and symptoms of both acromegaly and hyperprolactinemia.27 The diagnosis of this group of adenomas requires more complex IHC and ultrastructural analysis, and their differentiation is essential because it has clinical and prognostic implications. Morphologically, three subtypes may be identified: (1) mixed adenomas of cells secreting GH and cells secreting PRL, (2) somatotroph cell adenomas, and (3) acidophilic stem cell adenomas.27,30 In the first subtype, IHC shows labeling for GH and PRL with various degrees of intensity and distribution. At the ultrastructural level, two separate cell populations are seen. Somatotroph cell adenomas are rare tumors (less than 2% of all PAs and approximately 8% of tumors associated with acromegaly31,32) in which IHC shows labeling for both GH and PRL in the same type of tumor cell. Ultrastructural analysis shows a well-differentiated adenoma consisting of a population of monomorphic cells having characteristics of GH- and PRL-secreting cells. Acidophilic stem cell adenomas are very rare27 and their diagnosis has great clinical importance because they may be confused with prolactinomas once most patients show characteristics of hyperprolactinemia.33 Histologically they are chromophobic tumors, with focal oncocytic changes in cytoplasms. IHC shows labeling for PRL and, to a lesser extent, for GH in the cytoplasm of the same tumor cells. Electron microscopy is required for the accurate identification of these adenomas,33 and may reveal megamitochondria responsible for the oncocytic appearance in light microscopy.
ACTH-secreting adenomasACTH-secreting adenomas associated with Cushing's disease account for approximately 10%–15% of all Pas.34 Histologically, papillary formations are common, and strong labeling for ACTH may often be seen with the histochemical PAS technique and with IHC. Peripheral hyaline bundles, giving a “target cell” appearance, may sometimes be seen. These changes are called Crooke hyaline degeneration and correspond to the accumulation of intermediate cytokeratin filaments (IHC for cytokeratin shows this intracytoplasmic accumulation). This appears to be a direct effect of high serum cortisol levels on these pituitary cells.35
Gonadotropin-secreting adenomasGonadotropin-secreting adenomas (secreting FSH and LH) account for approximately 20% of all Pas.36 They do not usually cause a clinical syndrome related to hormone overproduction and are clinically characterized as non-functioning adenomas. Histologically, tumor cells are arranged in a diffuse growth pattern, but papillary structures frequently form around blood vessels,36 resulting in a pattern that mimics the formation of perivascular pseudorosettes. The use of monoclonal antibodies specific for β-FSH (the most common36), β-LH, and alpha-subunit (α-SU) is recommended for characterization because these lesions may show different degrees of reactivity for one or more gonadotropin units. The characterization of these adenomas by electron microscopy may be of scientific interest, but does not affect the clinical management of these patients.
TSH-secreting adenomasTSH-secreting adenomas are the least common PAs37 (less than 1% of all adenomas). IHC usually shows variable positivity for β-TSH, and usually for α-SU, also. Diagnosis may be difficult if the clinical presentation and immunoreactivity for TSH are not convincing. If this occurs, electron microscopy is mandatory for adequate diagnosis.
Silent adenomasThere are some clinically non-functioning PAs in which, despite the absence of the clinical syndrome or hormone hyposecretion or hypersecretion, an IHC labeling pattern and an ultrastructural appearance consistent with a secretory adenoma are seen. These are the so-called silent adenomas. Among these, those with the most significant clinical implications are “silent” corticotroph adenomas, characterized by immunoreactivity for ACTH (with an absence of any clinical signs of Cushing's disease or serum levels reflecting excess ACTH secretion). These adenomas characteristically show a high trend to bleeding and apoplexy (defined as the sudden occurrence of symptoms such as severe headache, nausea, vomiting, vision loss, cranial nerve palsy, and impaired consciousness with radiographic evidence of hemorrhagic infarction, often followed by hypopituitarism38), occurring in approximately one third of patients.39,40
Plurihormonal adenomasPlurihormonal adenomas are rare and show an unusual immunoreactivity for multiple pituitary hormones which are not related through cytogenesis or normal development of the anterior pituitary gland.41
Null cell adenomasApproximately 20% of PAs show no clinical or IHC evidence of hormone production.14,42 These tumors are called null cell adenomas, mainly because of the absence of ultrastructural characteristics providing specific differentiation. Histologically, oncocytic cell changes may be seen in some of these cases, and those adenomas may therefore be called oncocytomas.42 There is substantial overlap between null cell and gonadotroph adenomas, as some of these have been found to show weak, focal immunoreactivity for glycoprotein hormones. However, the differentiation of these two adenomas is of little clinical significance for patient management.14
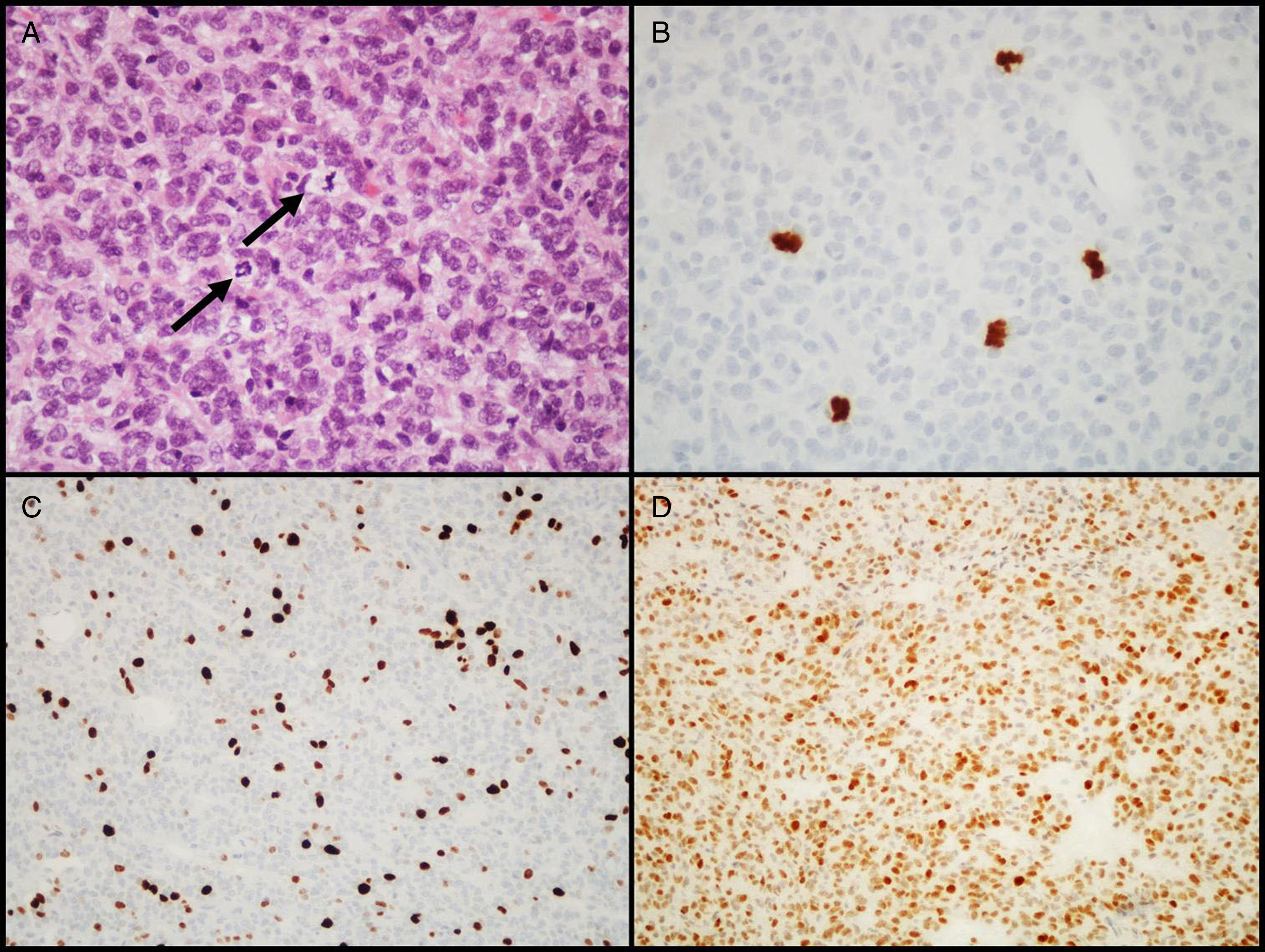
Atypical adenomasIn 2004, the WHO introduced the designation of atypical adenoma for tumors having histological characteristics suggesting aggressive clinical behavior (such as invasive growth).18 These tumors are characterized by a high mitotic index, a cell proliferation index (Ki-67) higher than 3%, and extensive immunopositivity for the p53 protein18 (Fig. 3). This latest classification of the WHO was somewhat controversial because differences between “typical” and “atypical” PAs were not clearly defined, as no cut-off values were established for criteria such as the number of mitoses or the percent positive nuclei and intensity of IHC labeling for the p53 tumor suppressor gene. Some authors have already suggested changes for future editions.43–45
. (A) Several mitotic figures (arrows) are seen in a high power field (HE, 400×). (B) Immunohistochemical confirmation of mitoses with the PHH3 antibody (PHH3, 400×). (C) Tumor shows a high proliferation index (8%, Ki-67, 200×). (D) Extensive nuclear immunoreactivity for p53 (p53, 200×).")
Diagnostic characteristics of atypical pituitary adenomas (example of an atypical null cell adenoma). (A) Several mitotic figures (arrows) are seen in a high power field (HE, 400×). (B) Immunohistochemical confirmation of mitoses with the PHH3 antibody (PHH3, 400×). (C) Tumor shows a high proliferation index (8%, Ki-67, 200×). (D) Extensive nuclear immunoreactivity for p53 (p53, 200×).
Other transformation markers of PAs have been proposed, including cathepsin B or metalloprotease-9 (MMP-9), the assessment of proliferation activity using antiapoptotic markers such as bcl-2, the analysis of DNA topoisomerase II-alpha indices, cyclooxygenase II expression, the detection of telomerase expression, or studies with galectin-3. Unfortunately, none of them has been shown to be more useful as a marker of biological behavior than the histological subtype based on hormone content and cell structure, which continues to be the best independent predictive marker of aggressive behavior. The absence of the p53 gene, a decreased expression of folliculostellate cells, the nm23 gene, p27 and p21 abnormalities, the analysis of vascular endothelial growth factor, CD34, fibroblast growth factor receptor 4, the pituitary tumor transforming gene, chromosome 11 deletions, and the microRNA profile have also been proposed as ways to assess the aggressiveness of these tumors, but the classification criteria for PAs have not been considered to date.46–48 Closer monitoring of patients with these types of adenomas is recommended.
Pituitary carcinomasPituitary carcinomas are very rare,2,49 accounting for less than 1% of all pituitary tumors.50,51 They are defined by the presence of cerebral, medullary and/or systemic metastases. There are no morphological criteria for differentiating a locally aggressive adenoma from a carcinoma when the tumor is limited to the sella turcica. The development of a pituitary carcinoma from an adenoma is exceptional, and data on this sequence are currently lacking.52 Most pituitary carcinomas are hormonally active invasive macroadenomas, and prolactinomas and ACTH-secreting tumors account for two thirds of them.50 Unlike PAs, carcinomas always show IHC overexpression for the p53 protein.50,53 Once malignancy is confirmed, prognosis is uncertain, with one-year survival in two thirds of patients.54
Spindle cell oncocytomasAs these tumors have non-specific clinical and neuroimaging characteristics, diagnosis is mainly based on their histopathological features. They are clinically indistinguishable from non-functioning adenomas, and patients may show signs and symptoms of hypopituitarism and vision disturbances.55
Histologically, as their name implies, they are characterized by a fusiform and oncocytic appearance of the cells. Unlike PAs, they show no immunoreactivity for neuroendocrine markers and pituitary hormones. Tumor cells are immunoreactive for the epithelial membrane antigen, vimentin, bcl-2, S-100, and galectin-3; they do not usually express or only focally express glial fibrillary acidic protein.55–58 The relatively recent observation of the thyroid transcription factor (TTF-1) in these tumors, and in other normal pituicytes and other tumors of the pituitary region, including pituicytomas and granular cell tumors of the neurohypophysis, suggests the possibility that they may all represent different variants derived from a common pathogenetic origin,59,60 and that the origin of these tumors are not folliculostellate cells of the anterior pituitary as originally thought.57
Tumors of the neurohyphohysisPituicytomasPituicytomas (formerly known as astrocytomas of the anterior pituitary gland or infundibulomas) are rare tumors; their clinical behavior has not yet been clearly elucidated. They appear to behave as tumors with a low degree of malignancy in most cases, with a certain trend to recurrence after subtotal resection.61
Morphologically, they consist of elongated piloid cells arranged in bundles, in a pattern that resembles a pilocytic astrocytoma (however, unlike pilocytic astrocytoma, most pituicytomas lack the biphasic pattern, the characteristic Rosenthal fibers, and eosinophilic granular bodies). In IHC, pituicytomas show no immunoreactivity for neuroendocrine markers or pituitary hormones. Tumor cells are typically positive for vimentin and protein S-100, and are immunoreactive for bcl-2 and TTF-1.62 While most pituicytomas express glial fibrillary acidic protein, labeling may be variable and even absent.56,57,59
Granular cell tumorsGranular cell tumors are rare (there are approximately 60 cases reported in the literature63), and are usually found incidentally in adult autopsies. They are generally slow-growing benign neoplasms, but a more aggressive clinical behavior has been reported in some cases.63
Histologically, these tumors consist of big polygonal cells with abundant granular cytoplasm (strongly positive for PAS with diastase), round nuclei with delicate chromatin, and uniform nucleoli. In IHC, most tumors are immunoreactive for NSE and CD68 and, as with pituicytoma and spindle cell oncocytoma, are also strongly positive for TTF-1.60 Unlike those arising in the peripheral nervous system, only a minority of those found in the sellar region are positive for S-100.63
Other lesions and tumors in the sellar regionMetastasesMetastases to the pituitary gland may be seen in 1% of surgical specimens, although the incidence of metastases found in pituitary glands at autopsy may be greater.8,64 Although up to 28% of tumors have been reported to cause pituitary metastases in autopsy series, most metastatic tumors are clinically asymptomatic.65 The posterior pituitary gland is affected more frequently than the anterior (occasionally with signs and symptoms of diabetes insipidus). Breast and lung cancer are the primary neoplasms most commonly causing pituitary metastasis.65
CraniopharyngiomasThese account for approximately 3% of all intracranial neoplasms and for approximately 10% of sellar region tumors.2,66 Most craniopharyngiomas occur in childhood and adolescence (from 5 to 15 years of age). In neuroimaging studies, craniopharyngiomas are typically calcified, solid, cystic (or solid-cystic) lesions with a complex lobar appearance.
Histologically, craniopharyngiomas show a complex, characteristic epithelial growth pattern, and are classified as grade I tumors according to WHO criteria.66 The WHO classification identifies two variants: adamantinomatous (characterized by stratified epithelium with basal cells arranged as a palisade, “wet” keratin formation, microcystic changes, and aberrant nuclear beta-catenin expression in up to 95% of cases) and papillary (characterized by simple non-keratinizing stratified pavement epithelium lining a connective tissue stroma, usually forming pseudopapillary structures),66 although these tumors may sometimes express both growth patterns in variable proportions.
Inflammatory lesionsPrimary inflammatory diseases of the pituitary gland are uncommon and may mimic sellar tumors. Hypophysitis has been classified into three categories based on its clinical and pathological presentation: (1) autoimmune lymphocytic hypophysitis (the most common and clinically relevant), (2) granulomatous hypophysitis, and (3) xanthomatous hypophysitis.67,68 Lymphocytes are not normally present in adenohypophysis, and a significant number of inflammatory cells in the pituitary gland therefore represent a pathological condition. Lymphocytic hypophysitis is an uncommon condition usually affecting women, particularly in the last part of pregnancy or in the postpartum period.69 It is very rare in men.67,68,70 It is thought to have an autoimmune basis, as both antibodies to pituitary cells and an association with other endocrine or immune diseases have been shown in approximately 20% of patients.69 Microscopically, lymphocytic hypophysitis is characterized by the lymphoplasmacytic infiltration of adenohypophysis, which in subsequent disease stages may cause the atrophy of gland parenchyma, a variable degree of fibrosis, and residual lymphoid aggregates.
Rarely, the pituitary gland may also be secondarily affected by systemic inflammatory or infectious processes (such as sarcoidosis or tuberculosis), which should therefore be ruled out before a final diagnosis of primary hypophysitis.
Other sellar area lesionsA great variety of other lesions and tumors may affect the pituitary gland and sellar region. These include tumors arising in the dura mater and sella turcica lining (such as meningioma), bone structures (such as chordoma), and bone marrow (such as plasmacytoma or Langerhans cell histiocytosis).
ConclusionsThe pathologist has a key role to play in the multidisciplinary team caring for patients with pituitary disease, and can only achieve an accurate diagnosis in close cooperation with the rest of the medical team.
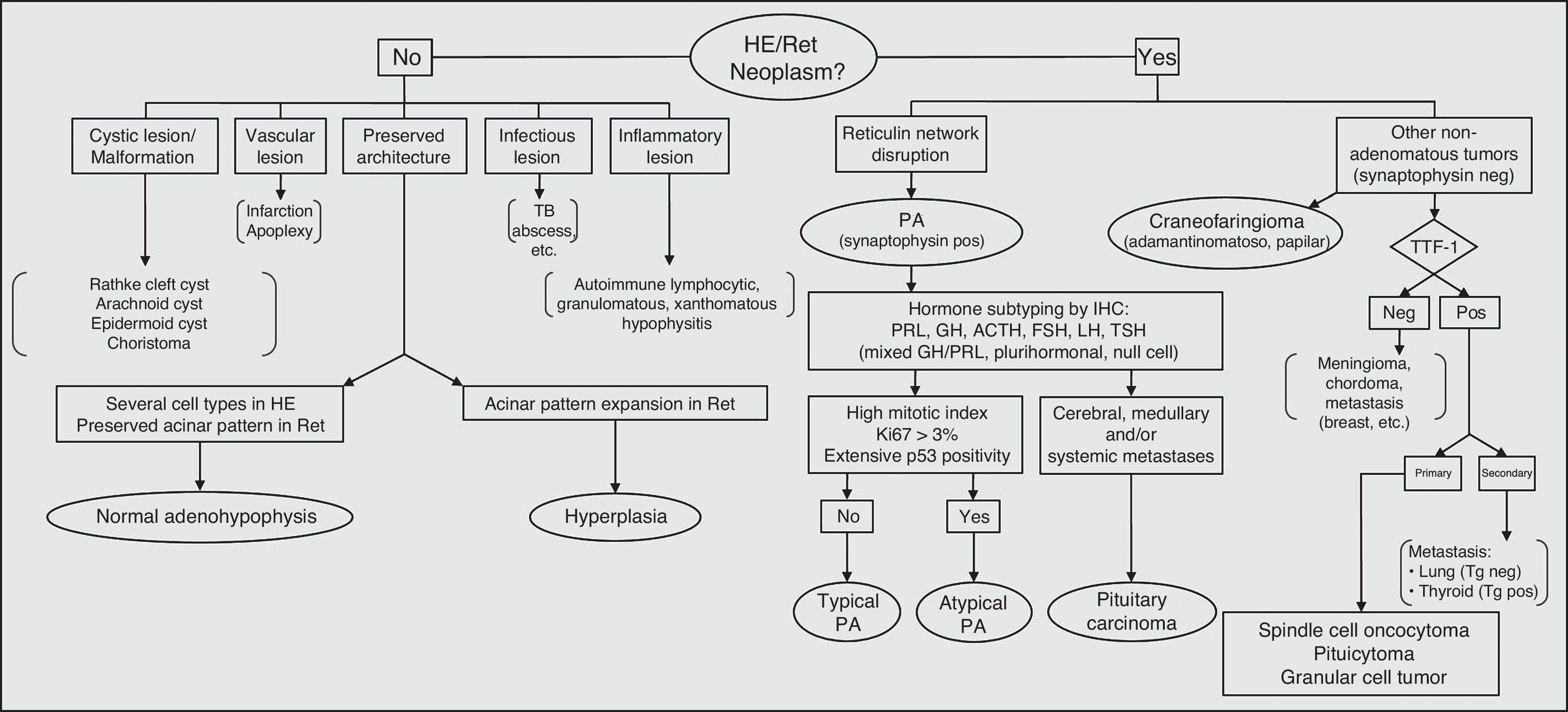
Fig. 4 shows a flow chart of the most common pituitary diseases including some cell differentiation IHC markers and prognostic markers. With new biomarkers for aggressive PAs and new data on genetic abnormalities associated with pituitary tumor pathogenesis, it is to be hoped that the relationship between typical and atypical adenomas and carcinomas is now clarified, thus allowing for greater diagnostic and prognostic accuracy.

Diagnostic algorithm in pituitary pathology. PA: pituitary adenoma; HE: hematoxylin–eosin; IHC: immunohistochemistry; neg: negative; pos: positive; Ret: reticulin; TB: tuberculosis; Tg: thyroglobulin; TTF-1: thyroid transcription factor.
- •
The hypophysis may be affected by a wide variety of lesions, some having similar clinical and radiographic characteristics. The possibility that the condition is not an adenoma should always be taken into account.
- •
Adequate pathological assessment of PAs requires extensive IHC and, in some cases, electron microscopy.
- •
Some PAs are intrinsically aggressive; the histological subtype based on hormone contents and cell structure continues to be the best predictive marker of aggressive behavior.
- •
The pathologist has a key role to play in the multidisciplinary team caring for patients with sellar region tumors.
The authors state that they have no conflicts of interest.
Please cite this article as: Tortosa F, Webb SM. Aspectos novedosos en histopatología de la hipófisis. Endocrinol Nutr. 2017;64:152–161.
