




Ectopic ACTH secretion (EAS) is an uncommon cause of ACTH-dependent Cushing's syndrome. Previous large studies1–3 concluded that two thirds of EAS tumors are located in the thorax and 8%–15% are located in the abdominal cavity. EAS tumors at other sites are less common; of note, up to 15% of tumors remain undetected. The adrenal glands are thus an extremely rare location.
Localization of these tumors can occasionally be difficult and may require extensive diagnostics test, therefore hypercortisolism normalization during this period is crucial. Adrenal steroidogenesis inhibitors as ketoconazole or metyrapone are preferred for their efficacy and safety. Somatostatin analogs (SSAs), have also been used to treat EAS with different results,4–6 and in some cases a paradoxical increase in ACTH and cortisol after SSAs treatment has been reported.7 In the present report, we describe a patient with adrenal EAS, who experienced a life-threatening worsening after conventional SSAs administration. We highlight the need to be aware of this rare presentation of EAS, and we remark the difficulties of EAS diagnosis and treatment.
A 48-year-old woman with newly diagnosed hypertension complained of amenorrhea, intense fatigue, muscular weakness and easy bruising, which had increased rapidly over the previous 3 months. Her past history was unremarkable except for recent treatment with enalapril. On examination the patient had normal body mass index, tanned skin with intense thinning with bruising, marked proximal limb muscle atrophy, and a mild moon face. She did not display buffalo hump, purple striae, or supraclavicular fat pads.
Laboratory findings revealed: 3-fold normal rate urinary free cortisol (UFC), (250μg/day per two times; reference range 12.8–82.5μg/day) and mild hypokalemia (3.3mmol/l; reference range 3.50–5.10mmol/l); lack of cortisol suppression after low-dose (1mg) dexamethasone (19.67μg/dl, reference value <1.8μg/dl). Basal plasma cortisol was 29.56μg/dl (reference range, 5.27–22.45μg/dl), and ACTH was 47.83pg/ml (reference range, 4.7–48.8pg/ml). Therefore, ACTH-dependent Cushing Syndrome diagnosis was established. Pituitary-centered magnetic resonance imaging (MRI) showed no evidence of pituitary adenoma, and cortisol levels were virtually unchanged after 8-mg dexamethasone suppression test (from 18 to 20μg/dl). The bilateral inferior petrosal sinus sampling (BIPSS) showed no significant central-to-periphery ACTH gradient, thus ruling out a pituitary origin of ACTH excess.
The results of neck, thorax, and abdomen 3-mm-sliced computed tomography (CT) scan and MRI were unremarkable. The somatostatin receptor scintigraphy (SSRS) and (PET)/CT localized only a slightly higher concentration of somatostatin receptors and glucose uptake in the left adrenal gland, compared with the right. As an ectopic source of ACTH located in the adrenal could be associated with pheochromocytoma and the patient had recent hypertensive state, we also performed urinary catecholamine determination, which was normal, and metaiodobenzylguanidine (123I-MIBG) scintigraphy, which showed increased uptake in the left adrenal.
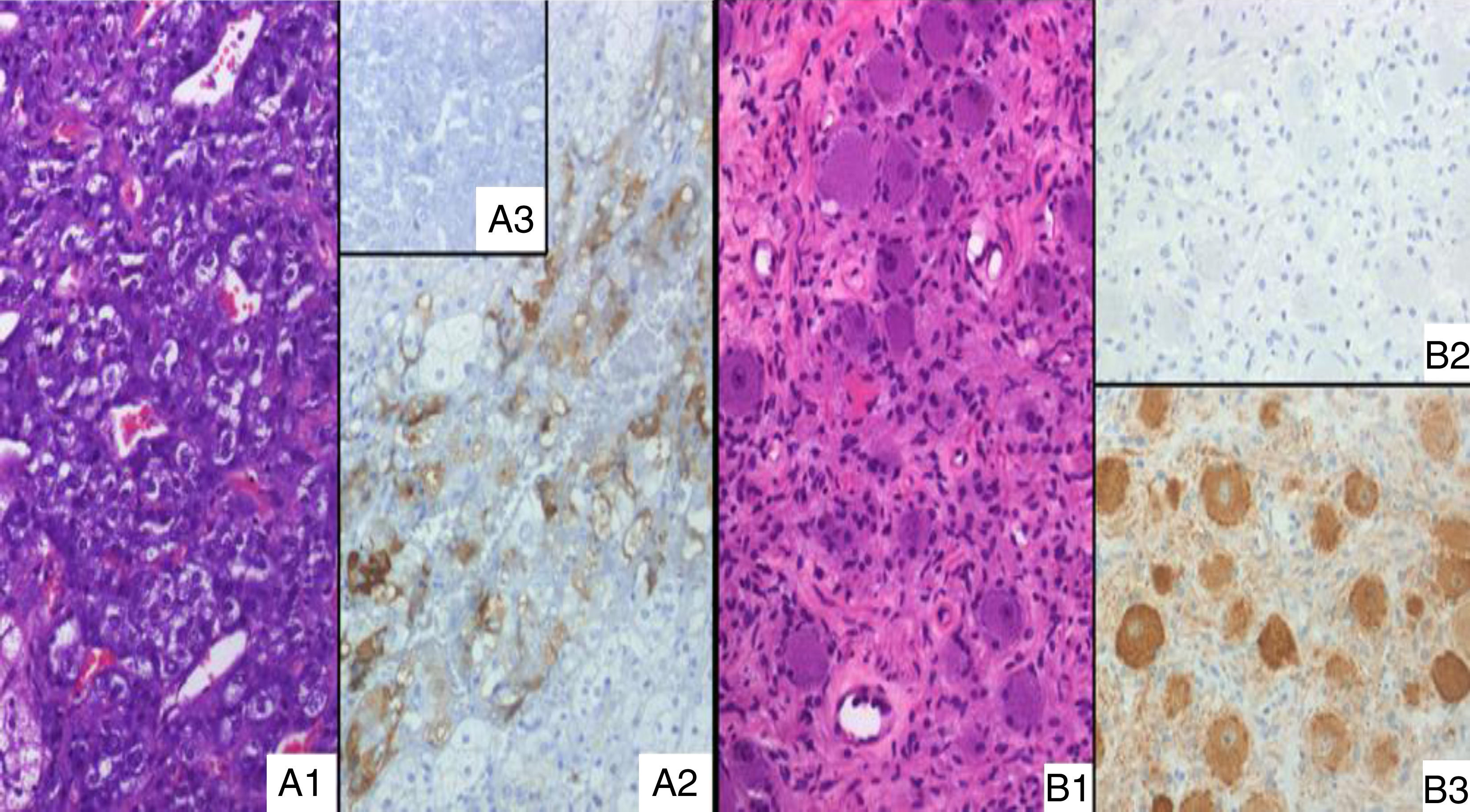
As SSRS imaging was positive, a high-dose extended-release SSAs was initiated (lanreotide 120mg) while waiting for ketoconazole provision; however, the patient's condition worsened and become life-threatening one week later, while UFC and ACTH raised up to 800μg/day and 102pg/ml respectively. Ketoconazole was started immediately (200mg three times daily) in a “block and replace” regimen. The patient underwent surgery; her left adrenal gland and a contiguous lesion, not detected previously, were removed. Immunohistochemical study revealed heterogeneous but unequivocal ACTH positivity in the medullary area of the adrenal, but not in other areas. This was interpreted as pathological, based on ACTH negative staining in three additional adrenal glands analyzed as controls (Fig. 1A). The other resected lesion turned out to be an extra-adrenal, 2×1.5cm ganglioneuroma; surprisingly, it had negative ACTH immunostaining (Fig. 1B). The patient's clinical condition has improved over the follow-up period and she was able to give up her hypertension pills. Her ACTH and urinary free cortisol levels remained normal at her latest control at ten months, but she still required cortisol replacement during stress.
 Adrenal medulla of the present case, without morphological abnormalities (H&E, ×20); (A2) positive ACTH immunostain of the same histological area as A1; (A3) absolute negative ACTH stain in an adrenal control case. (B1) Extra-adrenal ganglioneuroma (H&E, ×20); (B2) ACTH immunostain, absolutely negative; (B3) NSE positive expression in neuronal component, according to the diagnosis.")
(A1) Adrenal medulla of the present case, without morphological abnormalities (H&E, ×20); (A2) positive ACTH immunostain of the same histological area as A1; (A3) absolute negative ACTH stain in an adrenal control case. (B1) Extra-adrenal ganglioneuroma (H&E, ×20); (B2) ACTH immunostain, absolutely negative; (B3) NSE positive expression in neuronal component, according to the diagnosis.
EAS diagnosis is a challenge, and the patient's condition can be life-threatening during severe hypercortisolism. Despite clinical and biochemical evidence of ACTH-dependent Cushing's syndrome, up to 15% of tumors remain undetected.1 BIPSS is considered the gold standard for differential diagnosis between EAS and Cushing disease.2 An ectopic source of ACTH located in the adrenal gland occurs in less than 5% of cases, and is generally associated with pheochromocytoma or paraganglioma, not found in this case. We are no able to ensure that the high 123I-MIBG uptake was from the adrenal medulla, or from the contiguous ganglioneuroma; nevertheless, it is important to note that 123I-MIBG was associated with the highest percentage of false positives.8 Likewise, the sensitivity of SSRS in EAS is known to be relatively low, ranging from 25% to 60%,1,2,8 suggesting low receptor expression compared with other NETs. This could be an intrinsic characteristic of tumors causing EAS, though a functional mechanism involving selective downregulation of SSTR2 induced by elevated cortisol has also been advocated.9,10 Moreover, SSRS has been reported to become positive after successful glucocorticoid-antagonizing therapy.10 A recent systematic review9 analyzed various nuclear medicine imaging techniques in ectopic Cushing's syndrome and concluded that 68gallium-SSTR-PET/CT showed the highest SSTR5 affinity, and the highest sensitivity in EAS diagnoses. Unfortunately, this technique was not available in our center. Finally the same mechanism, selective downregulation of SSTR2, could be responsible for negative or paradoxical responses to traditional SSAs. Honestly, in this case we cannot completely rule out that the patient's deterioration merely was the natural course of the disease; however, we assume she had a paradoxical response on the basis of her dramatical worsening just after the SSAs administration, associated to an important rise in ACTH and UFC levels. In this sense, the use of the new SSAs pasireotide, with better SSTR5 affinity may be preferred for EAS treatment than, traditional SSAs: octreotide or lanreotide, which have mostly SSTR2 affinity.
In conclusion, EAS presents a major diagnostic challenge. Adrenal gland localization occurs in less than 5% of cases and is usually associated with ACTH-secreting pheochromocytoma, which was not found in our case. Surgery, if feasible, is the unique treatment option, with curative potential. To avoid the devastating effects of severe hypercortisolism, medical therapy must be initiated as soon as possible; Steroidogenesis inhibitors with fast and safe action must be the first medical treatment of choice. Others options like SSAs may have unpredictable responses.
Funding statementThis research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consentWritten informed consent was obtained from the patient. A copy of the written consent is available upon request for review by the Journal Editor.
Declaration of interestThe authors declare that they have no competing interests.
