



Classic pituitary apoplexy (PA) is an acute, life-threatening clinical syndrome caused by acute hemorrhage and/or infarction of the pituitary gland. PA is considered a neuroendocrinological emergency. However, there is no consensus about the best options for PA diagnosis and management.
ObjectiveTo develop a clinical practice guideline with a number of recommendations for diagnosis and treatment of patients with PA based on the medical evidence available, in order to help clinicians involved in their care.
MethodsThe clinical guideline for diagnosis and treatment of pituitary apoplexy issued in 2006 by the Neuroendocrinology Working Group of the Spanish Society of Endocrinology and Nutrition (SEEN) and the British Clinical Practice Guideline published in 2011 were taken as the basis. The text has been adapted to the format used in most international medical journals. For this, after updated medical literature, the quality of evidence and the strength of the recommendations were evaluated using the system proposed by the Agency for Health Care Policy and Research (AHCPR).
ConclusionsDiagnosis of pituitary apoplexy should be considered in all patients with acute severe headache with or without neuro-ophthalmic signs. Patients with PA must undergo a complete history and physical examination. All patients with suspected pituitary apoplexy should have urgent blood samples drawn to test electrolytes, renal function, liver function, coagulation screen, complete blood count, and basal levels of pituitary and peripheral hormones, and to rule out adrenocorticotropic hormone (ACTH) deficiency. Formal visual field assessment should be performed when the patient is clinically stable. Magnetic resonance imaging (MRI) is the imaging test of choice to confirm diagnosis. Indications for empirical urgent corticosteroid therapy in patients with PA include hemodynamic instability, impaired consciousness, reduced visual acuity, and severe visual field defects. In patients with these severe neuro-ophthalmic signs, surgery should be considered. Surgery should preferably be performed within seven days of the onset of symptoms. Patients with mild and stable signs may be managed conservatively with careful monitoring. Treatment and long-term follow-up of patients with PA should be conducted by a multidisciplinary team consisting, amongst others, of an experienced pituitary neurosurgeon, an ophthalmologist, and an endocrinologist.
La apoplejía hipofisaria (AH) clásica es un síndrome clínico agudo, potencialmente fatal, provocado por la hemorragia y/o infarto de la glándula hipofisaria. Constituye uno de los cuadros clínicos de urgencia neuroendocrinológica. Sin embargo, no existe un consenso claro sobre cuáles pueden ser las mejores opciones para su diagnóstico y tratamiento.
ObjetivoElaborar una guía de práctica clínica con una serie de recomendaciones para el diagnóstico y tratamiento de los pacientes con AH basadas en la evidencia médica disponible, que sirva de ayuda a los profesionales implicados en su cuidado.
MétodosSe ha tomado como base la guía clínica de diagnóstico y tratamiento de la apoplejía hipofisaria, publicada en el año 2006 por el Grupo de Trabajo de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición (SEEN), así como la Guía Británica de Práctica Clínica publicada en 2011. Se ha trabajado en la adaptación al formato utilizado en la mayoría de las revistas médicas internacionales. Para ello, tras la revisión bibliográfica actualizada, se ha evaluado la calidad de la evidencia y el peso de las recomendaciones de acuerdo con el sistema propuesto por la Agency for Health Care Policy and Research (AHCPR).
ConclusionesEl diagnóstico de apoplejía hipofisaria debería valorarse en aquellos pacientes con cefalea aguda grave, con o sin síntomas neurooftalmológicos. Se realizará una evaluación clínica completa. La evaluación analítica urgente incluirá medición de electrólitos, función renal y hepática, hemograma y estudio de coagulación y concentraciones basales de hormonas hipofisarias y periféricas. Será crucial descartar el déficit de hormona adrenocorticotropa (ACTH). Si el estado del paciente lo permite, debe realizarse una campimetría y una resonancia magnética (RM) urgente para confirmar el diagnóstico. En pacientes con inestabilidad hemodinámica, disminución de nivel de conciencia, disminución de la agudeza visual y defectos extensos en el campo visual se recomienda realizar descompresión quirúrgica en la primera semana tras el inicio de los síntomas. Los pacientes con sintomatología más leve pueden ser tratados de forma conservadora, bajo supervisión estrecha. El tratamiento de la AH debería realizarse en un centro donde exista disponibilidad y experiencia en el tratamiento neuroquirúrgico por vía transesfenoidal y posibilidad de seguimiento oftalmológico y endocrinológico.
Pituitary apoplexy (PA) is an acute, life-threatening clinical syndrome characterized by the sudden occurrence of headache, vomiting, visual changes with cranial nerve involvement, and decreased consciousness which is caused by pituitary gland hemorrhage and/or infarction. It is an endocrinological emergency. The diagnosis of PA requires a high level of suspicion, and different specialists should be involved in its management, including emergency physicians, neurosurgeons, ophthalmologists, and endocrinologists. There is however no clear consensus about the best option for the treatment of PA.
These clinical guidelines should not be considered as a clinical practice standard. They are only intended to provide a number of recommendations for the diagnosis and treatment of patients with PA based on the currently available medical evidence that will make it possible to harmonize as much as possible its management in both the acute phase and long-term follow-up and so improve the professional care of these patients. However, the level of evidence on which these recommendations are based is low, as no prospective and/or randomized studies are available. Such studies are needed to provide a higher level of evidence that will allow for further improving the treatment of these patients.
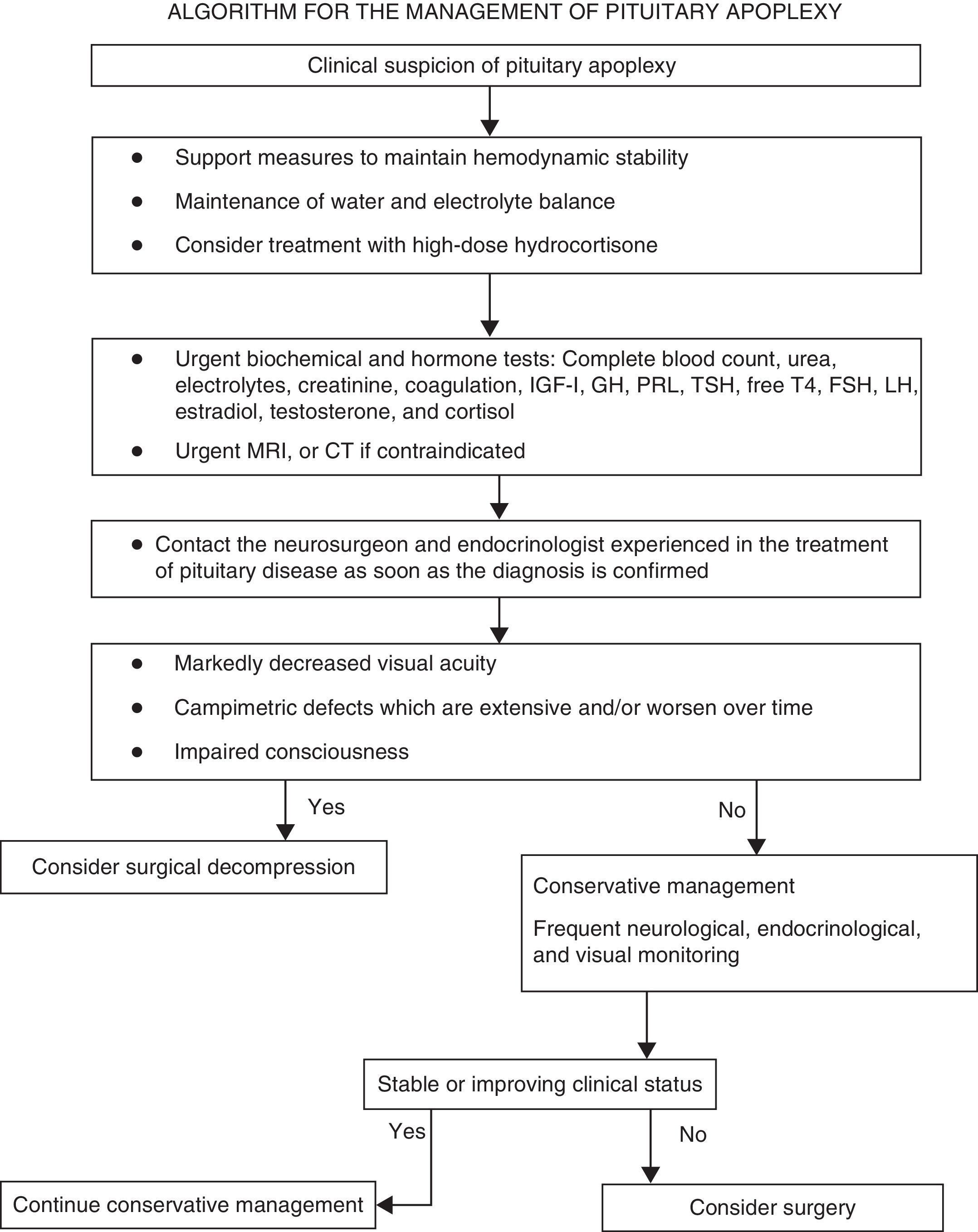
Summary of recommendationsRecommendations for the initial clinical assessmentClinical examination- •
A diagnosis of pituitary apoplexy should be considered in patients with severe acute headache, regardless of whether or not they have neuro-ophthalmic symptoms (√).
- •
The initial clinical assessment should always include a complete history aimed at detecting pituitary dysfunction symptoms, and a physical examination including the examination of the cranial nerves and a confrontation campimetry (√).
- •
If permitted by the clinical status of the patient, campimetry or perimetry (using a Humphrey visual analyzer or a Goldmann perimeter) should be performed within 24h of the start of the condition (√).
- •
All patients in whom pituitary apoplexy is suspected should immediately have samples taken to test electrolytes, kidney function, liver function, coagulation, complete blood count, and pituitary function (cortisol, prolactin, free thyroxine, TSH, IGF-1, GH, LH, FSH, and estradiol in women of childbearing age and testosterone in men (IV, C).
- •
All patients in whom pituitary apoplexy is suspected should urgently undergo magnetic resonance imaging (MRI) to confirm the diagnosis (III, B).
- •
Frequent monitoring of water and electrolyte balance, measures to maintain hemodynamic stability, and treatment with high glucocorticoid doses (III, B).
- •
Indications requiring the immediate empirical administration of glucocorticoids in patients with PA include hemodynamic instability, decreased consciousness, decreased visual acuity, and extensive visual field defects (IV, C).
- •
Hydrocortisone 100–200mg as a bolus, followed by 2–4mg/hour as a continuous IV infusion, should preferably be used (√).
- •
Once stabilization is achieved, patients should be transferred to a center where transsphenoidal neurosurgery and close ophthalmological and endocrinological monitoring may be performed (√).
- •
Patients with impaired consciousness and/or decreased visual acuity and/or acute, persistent, or gradually impaired severe visual field defects should undergo surgery (III, B).
- •
Surgery should preferably be performed within seven days of symptom occurrence (III, B).
- •
The procedure should be performed by a neurosurgeon experienced in transsphenoidal surgery on an elective basis.
- •
Patients with no or mild and stable neuro-ophthalmological symptoms and/or signs may be managed conservatively under close supervision (III, B).
- •
Neurological symptoms should initially be monitored every hour, but the interval may be lengthened to every four to six hours if the course of the disease is favorable and the patient is stable (√).
- •
Visual acuity and visual field defects should be examined daily until a clear trend to improvement is seen (√).
- •
Kidney function and electrolyte levels should be monitored every 24h or more frequently if needed (√).
In patients with impaired visual acuity or consciousness or a worsening of visual field defects, urgent MRI should be performed to plan for surgical decompression, including ventricular diversion in the event of hydrocephalus (IV, C).
Recommendations for clinical monitoring in the early postoperative periodClinical examination- •
During the first 24 or 48h after surgery, patients should closely be monitored to detect potential complications such as diabetes insipidus, vision loss, cerebrospinal fluid fistula, or adrenocorticotropic hormone/cortisol deficiency. Water balance should be recorded every hour (√).
- •
Visual acuity, eye movements, and confrontation campimetry should regularly be examined for the first 48h at the patient's bedside, and when possible, an instrumental examination should be performed (a Humphrey analyzer or a Goldmann perimeter) (IV, C).
- •
Kidney function and plasma electrolytes and plasma and urine osmolality should be assessed in the first 24 or 48h. These measurements should be performed at least once daily, and more frequently if required by the clinical status (IV, C).
- •
Cortisol levels should be measured at 09:00 AM on the second or third day after surgery in patients with no prior evidence of ACTH/cortisol deficiency. This will require the discontinuation of hydrocortisone in the evening before sampling (IV, C).
- •
In patients with ACTH/cortisol deficiency before surgery, treatment with hydrocortisone should be continued until the maintenance dose is achieved. These patients should be re-evaluated at 4 or 8 weeks to determine whether they will require long-term treatment (IV, C).
- •
Free thyroxine and TSH levels should be measured on the third or fourth day after surgery. If normal levels are found, they should be measured again at 4 or 8 weeks (IV, C).
- •
If visual impairment is found, urgent MRI should be performed and the patient should urgently be re-evaluated by the neurosurgical team (√).
- •
All patients who have experienced PA should be assessed 4 or 8 weeks after the acute episode. Pituitary function tests (baseline measurements and, when indicated, the relevant stimulation/suppression tests), cranial nerve examination, and campimetry and, optionally, optical coherence tomography (OCT) should be performed (√).
- •
All patients who have experienced apoplexy should be assessed annually using biochemical tests and pituitary function tests including cortisol, free thyroxine, TSH, LH, FSH, testosterone in men, estradiol in women of childbearing age, prolactin, IGF-1, and dynamic cortisol and GH tests if clinically indicated (√).
- •
Patients who have experienced apoplexy and have residual tumor will require radiographic follow-up (MRI) and, when indicated, should complete treatment with repeat surgery, medical treatment, or radiotherapy (III, B).
- •
Control MRI is recommended 3 or 6 months after apoplexy. If residual tumor or recurrence is found, monitoring is recommended every year during the first 3 or 5 years, and every 2 or 3 years thereafter (IV, C).
- •
At least annual monitoring is required in all patients. It is recommended that patients be followed up by a multidisciplinary team experienced in pituitary diseases (endocrinologists, neurosurgeons, ophthalmologists, and radiologists) (√).
These guidelines were prepared at the suggestion of the scientific committee of the Neuroendocrinology Group (GNE) of the Spanish Society of Endocrinology and Nutrition (SEEN) within the program for updating clinical practice guidelines in neuroendocrinology. In 2006, the Working Group of Neuroendocrinology of the SEEN issued the Clinical Guidelines for the Diagnosis and Treatment of Pituitary Apoplexy1. Based on this initial publication, and on the British clinical practice guidelines published on 2011,2 these guidelines have been updated and adapted to the format used in most international medical journals. To this end, after a literature review, the quality of evidence and the weight of the recommendations were assessed using the system proposed in 2004 by the Agency for Health Care Policy and Research (AHCPR), currently called the Agency for Healthcare Research and Quality (AHRQ)3.
Levels of evidence and grades of recommendation used (based on the system proposed by the Agency for Health Care Policy and Research)Levels of evidenceIa: Evidence obtained from meta-analyses of high quality clinical trials (controlled and randomized).
Ib: Evidence obtained from at least one high quality clinical trial.
IIa: Evidence obtained from one well-designed, controlled, nonrandomized study.
IIb: Evidence obtained from at least one well-designed, quasi-experimental study.
III: Evidence obtained from well-designed descriptive studies and case and control studies.
IV: Expert opinion.
- A
Level of evidence Ia or Ib.
- B
Level of evidence IIa, IIb, or III.
- C
Level of evidence IV.
√ Good clinical practice.
After a literature review of each of the different sections, a series of recommendations are given. The level of evidence appears in brackets after each recommendation, followed by the grade of recommendation.
DefinitionThe term PA, initially coined by Brougham et al. in 1950,4 is traditionally used to refer to an acute, life-threatening clinical syndrome characterized by the sudden onset of headache, vomiting, visual impairment, and decreased consciousness caused by pituitary hemorrhage or infarction. The syndrome was first described by Bailey5 in 1898. Syndrome definition is more clinical than pathological, because asymptomatic pituitary hemorrhage and/or infarction may be radiographic, surgical, or histopathological findings and should not be diagnosed as PA.6,7
PA may have two different clinical presentations:
- -
Acute PA. This is considered a life-threatening neuroendocrinological emergency that requires neurosurgical decompression in the vast majority of cases.6,7
- -
Subacute or subclinical PA. This has a slower, more torpid course and milder clinical signs and symptoms, is more common than the classical form and can be managed more conservatively, at least initially.7–9
The incidence of acute PA in pituitary adenomas ranges from 0.6% to 9% depending on the series,8,10–13 while the incidence rate of the subacute or subclinical form is up to 14–22%.10,12–14 PA affects more patients between the fifth and sixth decades of life, and is more frequent in males (1.6/1).11–15
Apoplexy is the first manifestation of pituitary adenoma in up to 80% of patients.7 It may occur in both functioning and non-functioning pituitary adenomas,11 but more commonly occurs in the latter due to the absence of a clinical hormone hyperfunction syndrome giving the alert about tumor existence. It has been reported to occur within non-adenomatous lesions such as craniopharyngiomas, pituitary cysts, and hypophysitis.8
In patients with PA, pathological study shows the presence of hemorrhagic infarction, bleeding, necrosis, or intratumoral cystic changes.13,16 Most series make no clear distinction between pure infarction and hemorrhage or hemorrhagic infarction. Some authors have reported that hemorrhage and hemorrhagic infarction are more frequently associated with the presence of a precipitating factor, a more severe clinical picture, and a poorer prognosis as compared to non-hemorrhagic infarction.7 However, small hemorrhagic areas, hemorrhagic infarction, or infarction have been found in imaging tests, during surgery, or in histopathological studies in up to 25% of pituitary macroadenomas,6,15,16 in most of which there were no apparent clinical signs. These cases cannot therefore be considered as PA.
PathophysiologyThe clinical signs of PA are caused by a rapid increase in the size of the intrasellar contents and the resultant increase in intrasellar pressure (median pressure of 47mmHg in PA versus 7–15mmHg under normal conditions), which causes the mechanical compression of optic tracts and internal cavernous sinus structures.17 The hemorrhage is usually encapsulated inside the tumor, but extravasation of blood into the subarachnoid space often occurs, leading to meningeal irritation symptoms. In large macroadenomas with suprasellar extension, obstructive hydrocephalus may occur as a complication of PA.
Lateral compression may affect cavernous sinus contents, causing oculomotor nerve palsy (ophthalmoplegia) in 70% of patients, with the third cranial nerve being most commonly affected.18 Upper optic chiasm compression causes visual acuity impairment and campimetric defects such as bitemporal hemianopsia in approximately 75% of patients.8,10,12 Lower optic chiasm compression may lead to the destruction of the sellar floor with cerebrospinal fluid fistula.17
A rapid and marked increase in intrasellar pressure will also cause ischemic necrosis of the anterior pituitary gland due to direct compression of the normal pituitary gland and decreased blood supply to the gland.17 Hypopituitarism will occur as a consequence of such destruction.19,20 Seventy-five to 90% of the gland must be destroyed for permanent hormone deficiencies to occur.17
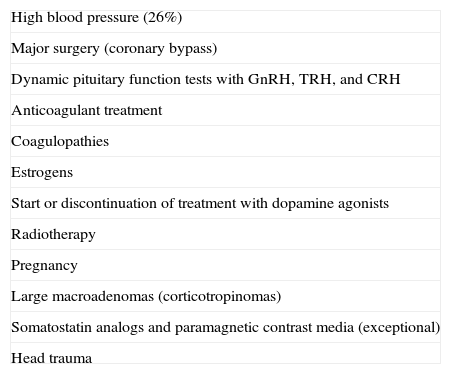
Precipitating factorsPrecipitating factors have been identified in up to 40% of cases of PA12 (Table 1). The most common factors include high blood pressure (26%),8,12 anticoagulation, and major surgery, especially coronary bypass surgery.21,22 In the latter, PA is mainly due to blood pressure fluctuations and the use of anticoagulant therapy.23
Precipitating factors in pituitary apoplexy.
| High blood pressure (26%) |
| Major surgery (coronary bypass) |
| Dynamic pituitary function tests with GnRH, TRH, and CRH |
| Anticoagulant treatment |
| Coagulopathies |
| Estrogens |
| Start or discontinuation of treatment with dopamine agonists |
| Radiotherapy |
| Pregnancy |
| Large macroadenomas (corticotropinomas) |
| Somatostatin analogs and paramagnetic contrast media (exceptional) |
| Head trauma |
Other promoting or precipitating factors include the dynamic tests used to assess pituitary function, such as those to measure GnRH, TRH, CRH, and insulin-induced hypoglycemia. Apoplexy occurred within two hours of the tests in 83% of patients, and within 88h in all patients.24
Anticoagulation, coagulopathies, the start or discontinuation of treatment with dopamine agonists10 or estrogens, radiotherapy,16 pregnancy,25 and head trauma may also induce the development of PA. Partial resection of a macroadenoma represents another risk factor because of compromised supply in the postoperative remnant.26
Adenomas secreting GH and ACTH, as well as large non-functioning adenomas, especially silent corticotropinomas, have been reported to be associated with a high risk of apoplexy27 (Table 1).
Clinical signsThe clinical picture usually evolves within hours or days7 depending on hemorrhage severity and volume, the presence of associated endocrine signs, and the parasellar structures involved. Acute PA with severe neurological deficiencies evolves in 24 or 48h and is considered as a life-threatening neuroendocrinological emergency that requires immediate surgical decompression.7,8 In subacute or “subclinical” PA, pituitary hemorrhage and/or infarction evolution may be slower and more insidious, with mild clinical signs.7–9
The most common symptom is headache, which is usually retro-orbital, although bifrontal or suboccipital headache may also be reported. The onset of headache is sudden and severe, is often associated with vomiting, and usually precedes all other symptoms.12,13,17,28
Visual involvement is common and occurs as oculomotor paresis. Symptoms of involvement of the 3rd cranial nerve are found in 50% of patients.10 Diplopia occurs in a high number of cases (67%).8 Variable visual acuity decrease (ranging from campimetric defects to bilateral amaurosis) and impairment consciousness may also occur (Fig. 1).

Seizures, hemiplegia due to brainstem compression, and diabetes insipidus have exceptionally been reported4,10 (Table 2).
As regards symptoms due to hormone dysfunction, almost 80% of patients have some deficiency of anterior pituitary hormones at the start of the condition, related to the presence of an undiagnosed microadenoma.29 ACTH/cortisol deficiency is found in 70% of patients. This is the most significant deficiency due to its attendant risk, given that acute adrenal insufficiency is the main cause of mortality and occurs as hemodynamic instability. GH, LH/FSH, and TSH deficiencies usually occur in more than 80%, 75%, and 60% of cases, respectively.11,12,18,19
Hyponatremia has been reported in 40% of patients, and is related to either inappropriate antidiuretic hormone secretion or hypocortisolism.8 Diabetes insipidus occurs less commonly.21
Prolactin levels may vary depending on the extent of pituitary ischemic necrosis. Some authors have suggested that normal or slightly elevated prolactin levels indicate a lower degree of gland destruction and, thus, a greater chance of pituitary function recovery after decompression surgery.17,19
DiagnosisAcute PA often mimics the clinical picture of other more frequent neurological emergencies, and initial diagnosis of the condition may therefore be difficult. Differential diagnosis should mainly be made with subarachnoidal hemorrhage and bacterial meningitis. Midbrain infarction (basilar artery occlusion) or cavernous sinus thrombosis, although less common, should also be considered.
Simultaneous or sequential occurrence of the following symptoms and signs will strongly suggest the presence of PA:
- 1
Sudden, continuous, predominantly retro-orbital headache.
- 2
Diplopia associated with visual field and/or visual acuity reduction.
- 3
Impaired consciousness.
MRI is the imaging test of choice because it confirms the suspected diagnosis in more than 90% of cases and allows for differential diagnosis with other neurological emergencies. MRI should be performed immediately in the event of visual impairment. Computed tomography (CT) only diagnoses less than 30% of PAs, but discloses the presence of a sellar mass in more than 80% of cases.11–14 In the initial phase, MRI usually shows a pituitary mass with hyperintense heterogeneous signal mainly in T1-weighted images and hypointense signal in T2-weighted images. Images of T2-weighted gradient echo sequences show the intratumoral hemorrhage in the acute phase as a very dark lesion that contrasts with adjacent structures. In subacute apoplexy, hemorrhage is seen in T2-weighted gradient echo sequence images as a dark ring.30
The test of choice to assess potential optic tract lesion is campimetry or perimetry. Although visual perimetry (a Humphrey visual analyzer or a Goldmann perimeter) allows for the objective assessment of visual field involvement, it is not helpful in establishing a prognosis about the course of campimetric damage. OCT is a simple examination that provides an image of the retina and optic disk and quantifies neuron loss at this level. OCT is very helpful for studying macular disease and has increasing applications in assessing neuron loss in the central nervous system, by extrapolating the findings in the retinal nerve fiber layer (RNFL) to macular and peripapillary level. This test may be very useful for the diagnosis and monitoring of patients with chiasm compression by pituitary tumors. Different studies have reported that the thinning of the RNFL at three months of surgical decompression is associated with a poor prognosis,27,31,32 although one study concluded that OCT findings do not always correlate to campimetric changes.33 Prospective studies are therefore needed to assess the prognostic value of OCT as a predictor of visual recovery after the treatment of patients with pituitary adenoma.
Recommendations for clinical assessment at the onset of pituitary apoplexyClinical examination- •
A diagnosis of pituitary apoplexy should be considered in patients with severe acute headache, regardless of whether or not they have neuro-ophthalmic symptoms (√).
- •
Patients diagnosed with pituitary adenoma should be warned of the possibility of apoplexy and its symptoms, particularly when they have precipitating factors (anticoagulant treatment, surgery with extracorporeal circulation, major surgery, etc.) (√).
- •
Clinical initial assessment must always include a complete history aimed at detecting pituitary dysfunction symptoms, and a physical examination including the examination of the cranial nerves and confrontational campimetry (√).
If permitted by the clinical status of the patient, campimetry or perimetry (using a Humphrey visual analyzer or a Goldmann perimeter) should be performed within 24h of the start of the condition (√).
Laboratory tests. Endocrinological assessment- •
All patients in whom pituitary apoplexy is suspected should immediately have samples taken to test electrolytes, kidney function, liver function, coagulation, complete blood count, and pituitary function (cortisol, prolactin, free thyroxine, TSH, IGF-1, GH, LH, FSH, and estradiol in women of childbearing age and testosterone in men (IV, C).
- •
In hemodynamically unstable patients, in whom support measures should be started immediately, blood samples for thyroid hormone (TSH, free thyroxine) and cortisol measurements should be taken before hydrocortisone IV is administered (IV, C).
- •
If samples cannot be directly sent to the laboratory at the time the patient is first seen, a serum sample should be stored for subsequent tests (√).
- •
All patients in whom pituitary apoplexy is suspected should urgently undergo MRI to confirm diagnosis (III, B).
- •
CT is indicted if MRI is contraindicated or cannot be performed (IV, C).
The treatment of PA is based on two therapeutic mainstays:
Medical treatment- -
This is based on the use of high-dose glucocorticoids, the control of water and electrolyte disorders, the maintenance of hemodynamic stability if required, and the treatment of hormone deficiencies.8,12,15
- -
The use of high-dose glucocorticoids is based on the presence of adrenal insufficiency and on their anti-inflammatory and antiedematous effects. Low plasma cortisol levels induce a lower vascular response to catecholamines, which promotes hemodynamic instability. They also increase vasopressin release, which results in decreased free water excretion, leading to hyponatremia. Glucocorticoid treatment should therefore be started in patients with hemodynamic stability and/or symptoms and signs of hypocortisolism before their cortisol levels are known.2,34
- -
The 2011 clinical guidelines2 advocate the use of hydrocortisone as an initial 100–200mg IV bolus, followed by a continuous infusion (2–4mg/h) or 50–100mg by the intramuscular (IM) route every 6 or 8h. The reason for this scheme is that the administration of hydrocortisone as IV boluses every 6 or 8h results in the saturation of cortisol-binding protein, so that a large amount of the hydrocortisone will be filtered in the kidney without exerting an effect. Potent long-lasting steroids (dexamethasone 2–16mg/day) are not considered a good option, but may be initially helpful to decrease edema if no early surgical decompression is performed. Mannitol may be added as needed.
- -
If improvement occurs after the acute episode, a rapid decrease in glucocorticoid dosage to oral hydrocortisone 20–30mg daily in three divided doses is recommended.2,34
- •
Frequent monitoring of water and electrolyte balance, measures to maintain hemodynamic stability, and treatment with high corticosteroid doses (III, B).
- •
Indications for the urgent empirical administration of glucocorticoids in patients with PA include hemodynamic instability, decreased consciousness, decreased visual acuity, and extensive visual field defects (IV, C).
- •
Hydrocortisone 100–200mg as a bolus, followed by 2–4mg/hour as a continuous IV infusion, should preferably be used. Alternatively, 50–100mg IM every 6h may be administered. Blood samples should first be taken for hormone and general laboratory tests as indicated in the laboratory test section. High-dose dexamethasone may initially be used in patients with cerebral edema (IV, C).
- •
In patients not meeting the above criteria, but with cortisol levels at 09:00h less than 550nmol/L (15μg/dL), steroids should also be administered with the same scheme (IV, C).
- •
Once stabilization is achieved, patients should be transferred to a center where transsphenoidal neurosurgery and close ophthalmological and endocrinological monitoring may be performed (√).
Surgical decompression, preferably by the transsphenoidal approach, is required. No agreement exists, however, as to whether surgery should always be performed or as to the time when it is most convenient.8,12,18 There are no prospective, randomized clinical studies on which recommendations may be based, and the currently available scientific evidence comes from retrospective studies and case reports. The transsphenoidal approach is preferred in most patients due to its low morbidity and mortality.
It should be stressed that, once stabilized, patients should be managed at hospitals where neurosurgical treatment is available, preferably at those with experience in transsphenoidal surgery,2,34 and where close ophthalmological and endocrinological monitoring may be performed. The British clinical guidelines for PA management consider surgeons who perform five or more procedures by the transsphenoidal approach every year to be expert.2
Most literature series advise urgent surgery in patients with impaired consciousness, hypothalamic involvement, the sudden onset of amaurosis, or decreased visual acuity.8,12,34,35 Surgery is also advised for patients with a worsening of campimetric defects and/or progressive impairment of visual acuity and/or neurological symptoms.8,12
Older retrospective series advocate decompression surgery in all cases8 based on the improvements in visual acuity, campimetric defects, ophthalmoplegia, and hormone deficiencies seen in most patients.28 Such improvements already occur in the early postoperative period and continue for several weeks after surgery.8 Different studies have reported that improvement is more complete if surgery is performed within seven days of the onset of symptoms.8,13,36,37
Recommendations for surgery- •
Patients with impaired consciousness and/or decreased visual acuity and/or acute, persistent, or gradually impaired severe visual field defects should undergo surgery (III, B).
- •
Surgery should preferably be performed within seven days of symptom occurrence (III, B).
- •
The procedure should be performed by a neurosurgeon experienced in transsphenoidal surgery on an elective basis. Urgent decompression by a neurosurgeon on duty call with no experience in this procedure should only be reserved for severe cases requiring immediate surgery (IV, C).
Recent retrospective studies6,38–40 report that patients with mainly oculomotor involvement, with no decreased consciousness, and with vision improvement in the first few days may be managed with conservative treatment alone. In these patients, the ophthalmological and endocrinological prognosis does not differ from that of patients treated with surgery. However, the bias involved in the fact that the patients undergoing surgery are usually those with severe symptoms at the start or with a poorer course after conservative management should be taken into account.
Another patient subgroup amenable to conservative management includes those with a milder clinical presentation (subclinical pituitary apoplexy).14,40
This situation especially applies to patients with prolactin-secreting adenomas. In these cases, treatment with a dopamine agonist not only controls prolactin levels, but also decreases tumor size.35
An overview has been made of the different criteria upon which decisions regarding initial treatment are taken. The form of presentation of the PA, its clinical course, a good response to glucocorticoid treatment, and the availability of an experienced neurosurgeon should all be considered.36 Some studies have proposed the analysis of radiographic characteristics.38,41 Uncontrolled studies have reported that the presence of a single hypodense area inside the tumor, as well as the early involution of the lesion, is often associated with the spontaneous resolution of the condition, which would support the maintenance of conservative management in these cases.38
A recently published study proposed the use of a scoring scale that allows for establishing PA severity, based on the Glasgow scale, visual acuity, the presence of visual field defects, and the degree of oculomotor palsy.42 This scale may represent a patient assessment tool based on objective data.
Recommendations for conservative managementPatients with no or mild and stable neuro-ophthalmological symptoms and/or signs may be managed conservatively under close supervision (III, B).
Neurological symptoms should initially be monitored every hour, but the interval may be lengthened to every four to six hours if the course of the disease is favorable and the patient is stable (√).
Visual acuity and visual field defects should be examined daily until a clear trend to improvement is seen (√).
Kidney function and electrolyte levels should be monitored every 24h or more frequently if needed (√).
Recommendations for surgery in patients on initial conservative management- •
In patients with impaired visual acuity or consciousness or a worsening of visual field defects, urgent MRI should be performed to plan for surgical decompression, including ventricular diversion in the event of hydrocephalus (IV, C).
- •
The decision to perform surgery or conservative management in patients with PA should be taken by a multidisciplinary team including a neurosurgeon, an endocrinologist, and an ophthalmologist (√).
- •
Treatment alternatives and the therapeutic modality decided upon should clearly be explained to patients, who should provide their informed consent if at all possible (√).
- •
Patients or their relatives should be given an explanation sheet containing simple and clear information about pituitary tumors and PA (√).
The postoperative care of patients who undergo surgery for PA is similar to that provided after pituitary tumor surgery.
Postoperative diabetes insipidus occurs in up to 16% of patients who undergo surgery for PA8. Other potential postoperative complications include cortisol deficiency, vision loss, cerebrospinal fluid fistula, and meningitis.
Adrenal and thyroid function should be assessed in the early postoperative period. Cortisol testing on a sample taken at 09:00h is the test of choice for the initial assessment of recently operated patients.43 Thyroid function (TSH and free thyroxine) should be tested on the third or fourth day after surgery and, if normal, should be assessed again at 6–8 weeks.
Visual acuity, campimetric defects, and oculomotor palsy improve in most patients after surgical decompression.8,10,11 Such improvement is seen in the early postoperative period and often continues weeks after surgery.44
Recommendations for clinical monitoring in the early postoperative periodClinical examination- •
During the first 24 or 48h after surgery, patients should be closely monitored to detect potential complications such as diabetes insipidus, vision loss, cerebrospinal fluid fistula, or ACTH/cortisol deficiency. Water balance should be recorded every hour (√).
- •
Visual acuity, eye movements, and confrontational campimetry should regularly be examined for the first 48h at the patient's bedside, and when possible, instrumental examination should be performed (a Humphrey analyzer or a Goldmann perimeter) (IV, C).
- •
Kidney function and plasma electrolytes and plasma and urine osmolality should be assessed in the first 24 or 48h. These measurements should be performed at least once daily, and more frequently if required by the clinical status (IV, C).
- •
Cortisol levels should be measured at 09:00 AM on the second or third day after surgery in patients with no prior evidence of ACTH/cortisol deficiency. This requires the discontinuation of hydrocortisone on the evening before sampling (IV, C).
- •
In patients with ACTH/cortisol deficiency before surgery, treatment with hydrocortisone should be continued until the maintenance dose is achieved. These patients should be re-evaluated at 4 or 8 weeks to determine whether they will require long-term treatment (IV, C).
- •
Free thyroxine and TSH levels should be measured on the third or fourth day after surgery. If normal levels are found, they should be measured again at 4 or 8 weeks (IV, C).
- •
If visual impairment is found, urgent MRI should be performed and the patient should be urgently re-evaluated by the neurosurgical team (√).
Different studies have shown that partial or total functional recovery of the anterior pituitary gland occurs in 50% of patients who have suffered PA.17
After PA, patients evolve in the following days or months to a state of partial or complete hypopituitarism which may be transient or permanent. Approximately 80% of patients will require some type of hormone replacement therapy. GH deficiency is most common (80–90%), followed by deficiencies in gonadotropins (60–80%) and ACTH/cortisol (60–80%), TSH (50–60%), and arginine vasopressin (10–25%).8,18,19,38
An improvement in visual signs after initial treatment often persists or even continues during follow-up. Recovery is less likely in patients who have experienced unilateral or bilateral visual loss.38 Diplopia is the symptom that remits first, while visual field reduction improves more slowly.30,44 OCT provides information not previously available in pituitary disease, is easy to perform, and has no contraindications. It should therefore be included in routine examination of these patients in the mid-term follow-up, because it may help determine the prognosis of visual field lesions.
The recurrence of apoplexy, as well as tumor growth or recurrence, has been reported in both patients on conservative management and those undergoing surgery.4,9,18,39 Long-term follow-up of all patients to detect potential recurrence is therefore needed.45
Recommendations for mid- and long-term follow-up- •
All patients who have experienced PA should be assessed 4 or 8 weeks after the acute episode. Pituitary function tests (baseline measurements and, when indicated, the relevant stimulation/suppression tests), cranial nerve examination, and campimetry and, optionally, OCT should be performed (√).
- •
All patients who have experienced apoplexy should be assessed annually using biochemical tests and pituitary function tests including cortisol, free thyroxine, TSH, LH, FSH, testosterone in men, estradiol in women of childbearing age, prolactin, IGF-1, and dynamic cortisol and GH tests if clinically indicated (√).
- •
Patients who have experienced apoplexy and have residual tumor will require radiographic follow-up (MRI) and, when indicated, should complete their treatment with repeat surgery, medical treatment, or radiotherapy (III, B).
- •
Control MRI is recommended 3 or 6 months after apoplexy. If residual tumor or recurrence is found, monitoring is recommended every year during the first 3 or 5 years, and every 2 or 3 years thereafter (IV, C).
- •
At least annual monitoring is required in all patients. It is recommended that patients be followed up by a multidisciplinary team experienced in pituitary disease (endocrinologists, neurosurgeons, and radiologists) (√).
Prospective and/or controlled and/or randomized studies are needed to answer the following questions:
- •
When is the best time for performing surgical decompression when it is required?
- •
Which patients will benefit from surgery as compared to conservative management? Can a clinical grading scale allowing for the differentiation of these patients be developed and validated?
- •
What is the long term neuro-ophthalmological and endocrinological prognosis of surgically treated versus conservatively managed patients?
The authors state that they have no conflicts of interest.
Please cite this article as: Vicente A, Lecumberri B, Gálvez MÁ, en nombre del Grupo de Trabajo de Neuroendocrinología. Guía de práctica clínica para el diagnóstico y tratamiento de la apoplejía hipofisaria. Endocrinol Nutr. 2013;60:582–582.
The names of the components of the Neuroendocrinology working group are related in Annex at the end of the article.
