


Diffuse large B-cell lymphoma is the most common histological subtype of non-Hodgkin lymphoma, accounting for approximately 25% of cases.1 It usually occurs as a rapidly growing mass associated with “B” systemic symptoms (fever, weight loss, and night sweats) in 30% of the patients.2 Serum lactate dehydrogenase (LDH) is increased in more than 50% of the patients. Primary lymphomas of the nervous system account for 6.6% of all primary cerebral tumors.3 Diffuse large B-cell lymphoma is an aggressive but potentially curable tumor.
A case of hypothalamic diffuse large B-lymphoma with an unusual course is reported below.
This was a 53-year-old female patient with high blood pressure, hypertensive heart disease, type 2 diabetes mellitus, dyslipidemia, and morbid obesity.
In 2008 she was admitted to the neurosurgery department for visual disturbances, polyuria, polydipsia, and drowsiness. Magnetic resonance imaging (MRI) showed a suprasellar lesion (craniopharyngioma versus meningioma). Treatment with dexamethasone 4mg every 8h was prescribed before surgery; a repeat MRI performed weeks later showed that the lesion had disappeared. During admission, the patient was diagnosed with central diabetes insipidus, and treatment with intranasal desmopressin was started. Evaluation of hypothalamic–pituitary function revealed ACTH, TSH, FSH, and LH deficiency. Replacement therapy was started with hydrocortisone 20mg and levothyroxine.
The patient was monitored at endocrinology outpatient clinics for the following four years. Annual imaging tests showed no lesion recurrence.
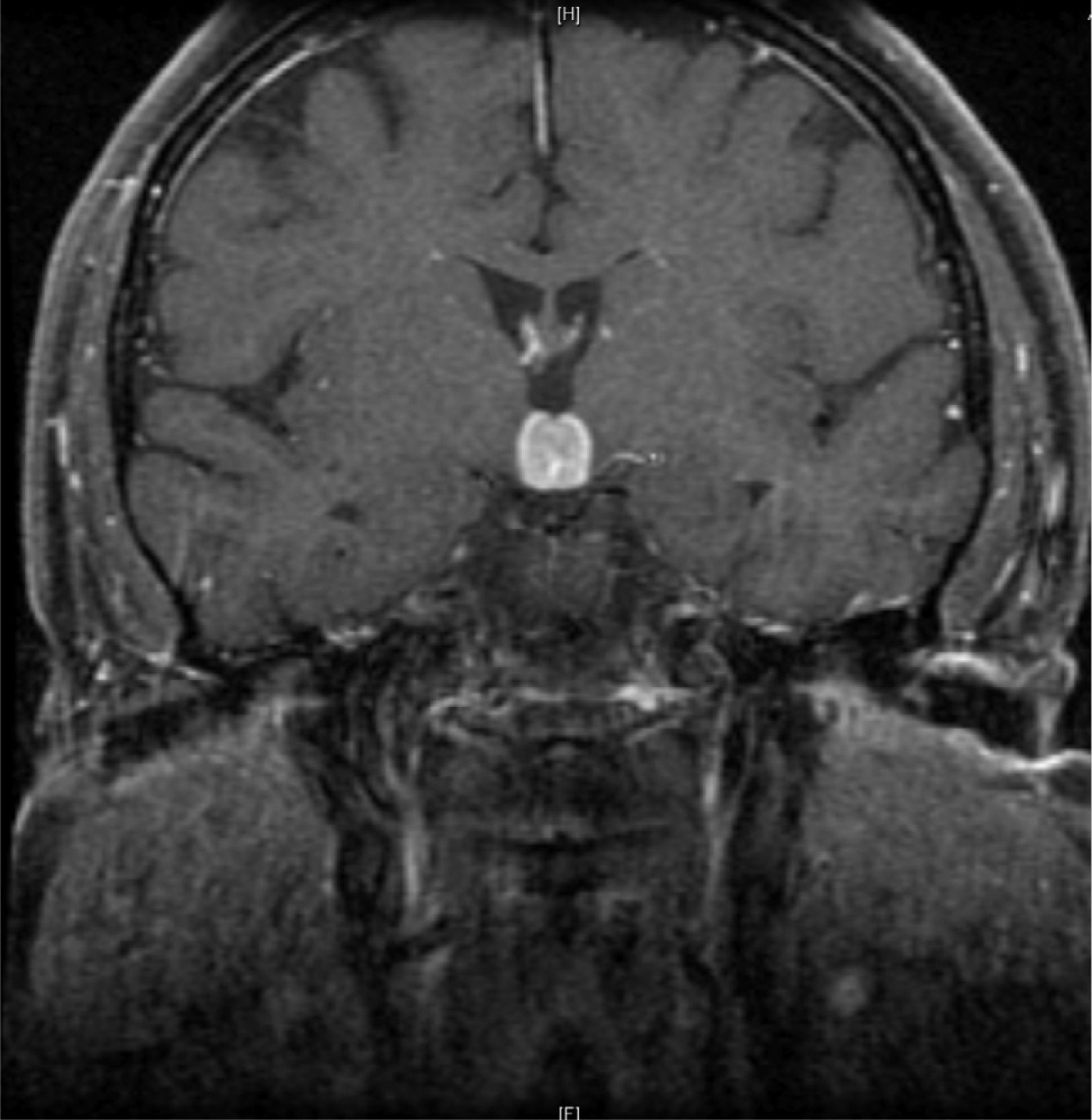
Initial neurological symptoms recurred in 2012, and MRI was therefore performed again (Fig. 1). Treatment with dexamethasone was started again, leading to disappearance of both symptoms and las lesions.

Clinical signs and symptoms recurred two months later. An MRI of the brain again showed a well-defined 16mm×15mm mass in the suprasellar region, enhanced by contrast and with a mass effect on the third ventricle, surrounded by perilesional edema. Based on the clinical signs and symptoms, images, and history of lesion remission with steroid treatment, hypophysitis,3 sarcoidosis,4 Langerhans cell histiocytosis,5 Erdheim-Chester disease,6 low-grade lymphoma, and other less probable conditions such as meningioma or demyelinating disease were considered in differential diagnosis. A complete blood count showed no changes, while blood chemistry results included: sodium, 145mg/dL (NR: 135–145); LDH, 646IU/L (NR: 230–480); and CRP: 34mg/dL (NR: 0–5). Angiotensin-converting enzyme was normal. Tests for autoimmunity (ANA, ANCA, anti-DNA and aquaporin antibodies) were negative. Results of hypothalamic–pituitary function tests included: cortisol, 2.87μg/mL (NR: 10–25); FSH, 0.862mIU/mL (VN: 3.5–21.5mIU/mL); LH, <0.1mIU/mL (NR:2.4–95.6mIU/mL); TSH, 0.037uIU/mL (NR:0.5–5); free T4, 9.2pg/mL (NR:8–18); PRL, 8.08ng/mL (NR: 1.9–25); IGF-1, 85ng/mL (NR: 55–360). Proteinogram, immunoglobulins, serology for syphilis, Borrelia and Brucella, immunocytology, and beta-2-microglobulin were normal. No changes were found in cerebrospinal fluid. A CT scan of the chest and abdomen, a gallium scan,6,7 and a whole body bone scan showed no relevant findings. Because of negative results in supplemental tests and lack of clinical response to repeat steroid treatment, lesion was biopsied. Histological findings were consistent with diffuse large B-cell lymphoma, positive for CD20 and DC79, with a proliferation index Ki-67>80%.
Primary central nervous system lymphomas are usually diagnosed in people aged 45–70 years, with a mean age at diagnosis in the fifth decade, as occurred in our patient.7 Symptoms at diagnosis included headache, blurred vision, motor problems, and cranial nerve changes. The parasellar location is extremely rare, and few cases have been reported in the literature. Most of these were B-cell lymphomas, of which approximately 40% were diffuse large B-cell lymphomas. Hypothalamic–pituitary dysfunction is common at diagnosis.8 Standard combination chemotherapy, helpful for the treatment of systemic lymphomas, is ineffective. Treatment of choice is usually methotrexate, or radiation therapy if this drug fails.9
Urine could not be alkalinized in our patient, and she was therefore not treated with methotrexate because of its nephrotoxicity. After several radiation therapy courses, she had a favorable response, with a significant decrease in lesion diameter. Panhypopituitarism, present since the initial episode, was not reversed.
The unique characteristic of our case was hypothalamic location of the lesion, as well as the remission time after corticosteroid therapy, up to four years. Initial response to corticosteroids has been reported in cerebral lymphomas in up to 70% of the cases. However, clinical and radiographic improvement is usually transient, and the disease tends to recur a few months after drug discontinuation.10 Although our patient was only sporadically treated with dexamethasone when symptoms occurred, the initial remission was sustained for four years.
Please cite this article as: López Navia AM, Galván Díaz B, Tejera Pérez C, Hernández Lavado R, Morales Pérez F. Lesión hipotalámica evanescente por linfoma B difuso de células grandes. Endocrinol Nutr. 2014;61:335–336.
