


La osteogénesis imperfecta (OI) es un trastorno genético caracterizado por fragilidad ósea debido generalmente a una mutación en los genes COL1A1 o COL1A2 que codifican para la cadena α del colágeno tipo 1, la principal proteína del hueso1–4.
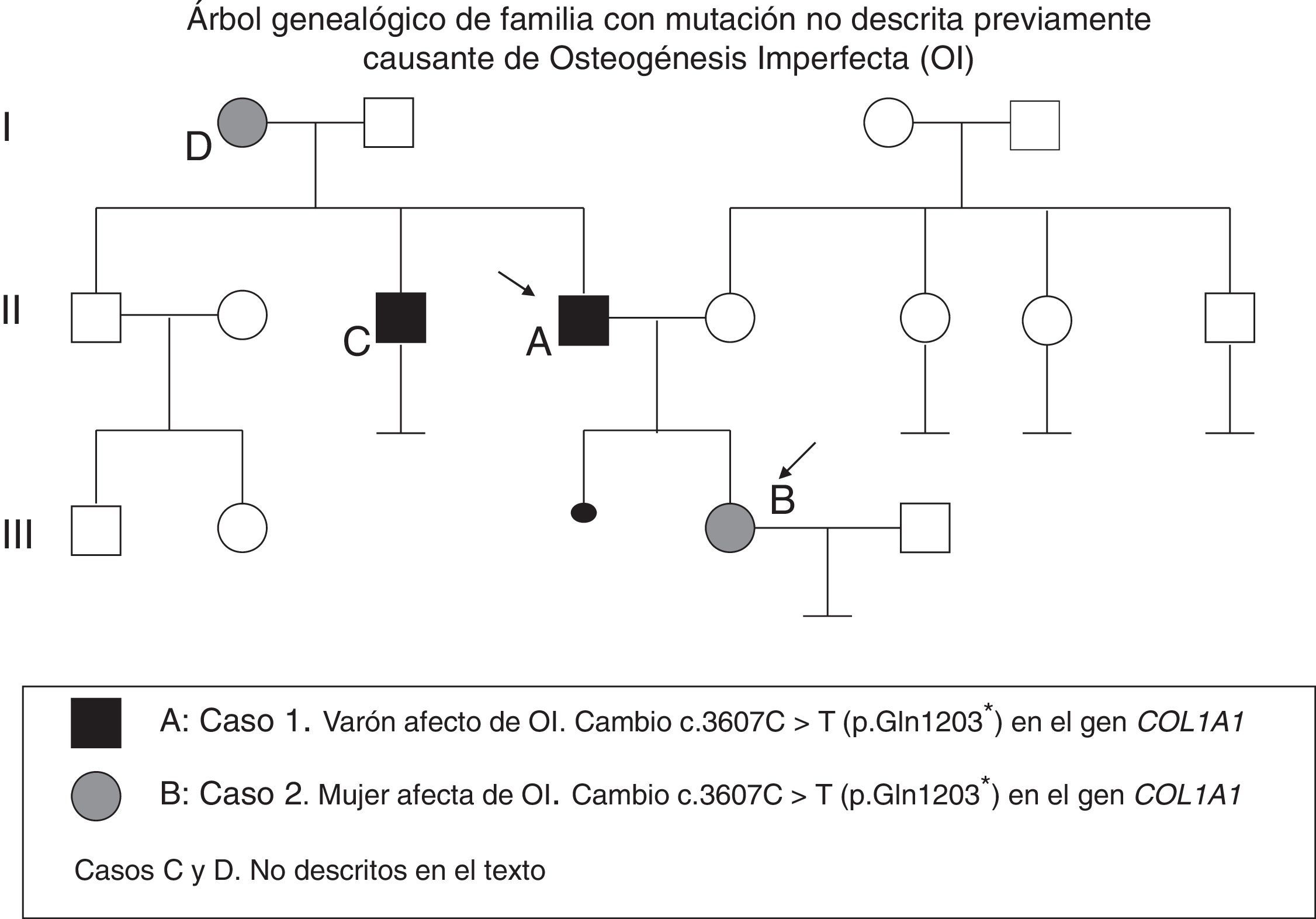
Describimos 2 pacientes afectados de OI (padre e hija), cuyo estudio genético reveló un cambio c.3607C>T (p.Gln1203*) en el gen COL1A1 no descrito hasta ahora. Revisamos las posibles opciones reproductivas.
Caso 1Varón de 55 años diagnosticado de OI tras nacer su hija afectada (fig. 1). Ha presentado al menos 10 fracturas hasta los 14 años y varias luxaciones. Ha sido diagnosticado de sordera mixta. Exploración: talla 158cm, escleras azules, dientes normales, no presenta deformidades óseas. Analítica: calcio, fósforo y creatinina normales, 25OH-vitamina D: 16ng/ml, con PTHi: 65.,7pg/ml (VN: 15-65) y calciuria en 24h: 260mg.
.")
Densidad mineral ósea medida por densitometría (DEXA): T-score medio en columna lumbar (CL) –2,2 (–3,7 en L4). Se recomendó tratamiento con alendronato más calcio y vitamina D con buena respuesta (T-score en CL tras 4 años de tratamiento –0,9).
El estudio genético reveló mutación en gen COL1A1: c.3607C>T (p.Gln1203*).
Caso 2Mujer de 21 años, hija del paciente número 1, diagnosticada de OI al nacimiento. Durante su gestación se detectaron tibias incurvadas y retraso del crecimiento intrauterino (peso al nacer: 2kg). Presentó su primera fractura a los 4 días de vida, y varias posteriormente, sobre todo en los miembros inferiores, hasta los 16 años.
A los 7 años la DEXA mostró una Z score –2,83 en CL; recibió pamidronato intravenoso y posteriormente alendronato oral hasta los 14 años, con ascenso de Z score en CL hasta +1,2, manteniéndose normal desde entonces. Exploración: talla 157cm, escleras azules, dientes normales, no presenta deformidades y la audición es normal.
Por su preocupación sobre la forma de herencia de su enfermedad y sus opciones con vistas a una futura maternidad, se hizo estudio genético dirigido, que confirmó que es portadora de la mutación detectada previamente en su padre. Se realizó asesoramiento genético.
La fragilidad ósea es la característica principal de la OI, determinando múltiples fracturas sin traumatismo o con traumatismo mínimo, y deformidades progresivas en los casos más graves. Otros signos son talla baja, escleras azules, dentinogénesis imperfecta e hipoacusia en adultos.
El diagnóstico suele ser precoz. En niños y adolescentes los bifosfonatos intravenosos han demostrado eficacia en reducir fracturas, mejorar el dolor, la movilidad y talla final. Sin embargo, muchos pacientes llegan a adultos sin diagnóstico. La mayoría de los adultos afectados tienen osteoporosis, siendo el tratamiento de elección los bifosfonatos5–7.
Generalmente la OI es causada por mutaciones en genes que codifican para las cadenas α1 y α2 del pro-colágeno tipo 1 (genes COL1A1 y COL1A2) con herencia autosómica dominante, aunque también se han identificado otros genes implicados como CRTAP y LEPRE-1, que dan lugar a formas infrecuentes de enfermedad con herencia autosómica recesiva. Al menos se conocen otros 11 genes relacionados: OSX, SERPINH1, PPIB, FKBP10, BMP1, CREB3L1, IFITM5, SERPINF1, SPARC, TMEM38B y WNT13,4,8.
Se han descrito más de 1.500 mutaciones diferentes. En nuestros pacientes se detectó mutación en el gen COL1A1: c.3607C>T (p.Gln1203*), que supone un cambio de aminoácido e glutámico por un codón de parada en la posición 1203 que genera una proteína truncada. Este cambio nucleotídico no ha sido descrito previamente en la bibliografía como asociado a OI, pero dado el efecto deletéreo que produce en la proteína es probable que sea su causa.
La mujer fue informada de la forma de herencia dominante y del riesgo de transmisión de su enfermedad (50% en cada embarazo). En los casos en los que se logra identificar la alteración el asesoramiento genético se completa con información sobre las opciones reproductivas9,10. En este caso las alternativas son:
Diagnóstico genético preimplantacional (DGP): tras un tratamiento de fertilización in vitro permite testar genéticamente los embriones y seleccionar los sanos. Es necesario conocer la mutación responsable de la enfermedad en esa familia, y obtener, mediante biopsia embrionaria, uno o 2 blastómeros del embrión. Se realiza diagnóstico genético molecular buscando la mutación conocida en los embriones y los no afectados son seleccionados para su transferencia al útero, lo que permite tener un hijo sin la enfermedad genética. El DGP es posible en España desde la aprobación de la Ley 14/2006, de 26 de mayo, sobre técnicas de reproducción humana asistida, y está incluido dentro de las prestaciones del Sistema Nacional de Salud. El principal argumento a favor de la realización del DGP es que evita el trauma del aborto y disminuye el estrés asociado con la espera del resultado del diagnóstico prenatal. En su contra supone un proceso largo, con un grado de efectividad bajo (15-20%) y elevadas tasas de embarazo múltiple, la posibilidad de manipulación de los embriones, la seguridad aún debatida de la biopsia de embriones y el alto coste.
Fertilización in vitro con óvulos de donante: sustituyendo el gameto del progenitor afecto por un gameto sano anónimo. Óvulos y espermatozoides son fecundados fuera del útero y el embrión es transferido al útero de la madre donde se implantará, libre de enfermedad.
Concebir un hijo y realizar un diagnóstico prenatal con técnicas como la biopsia de corion o la amniocentesis genética, que permiten obtener células fetales sobre las que dirigir estudios genéticos de OI. Si el feto porta la mutación no existe cura, tan solo la posibilidad de un aborto terapéutico.
Procedimientos no invasivos, como la detección de ADN circulante fetal en sangre materna, resultan prometedores.
Otras opciones de maternidad son el embarazo natural asumiendo riesgos o la adopción. La paciente está valorando pros y contras de estas alternativas.
Otra preocupación de mujeres con OI que reciben tratamiento con bifosfonatos durante la edad fértil es su seguridad en relación con futuras gestaciones, dado que permanecen durante años en la matriz ósea y traspasan la barrera placentaria. La supresión del recambio óseo podría ocasionar hipocalcemia fetal. Los datos en mujeres son escasos, pero en los casos descritos no se presentaron efectos adversos graves11. Está por establecer cuál es el periodo de seguridad.
