




Endocrine paraneoplastic syndromes are distant manifestations of some tumours. An uncommon but increasingly reported form is tumour-induced osteomalacia, a hypophosphatemic disorder associated to fibroblast growth factor 23 (FGF-23) secretion by tumours. The main biochemical manifestations of this disorder include hypophosphatemia, inappropriately low or normal tubular reabsorption of phosphate, low serum calcitriol levels, increased serum alkaline phosphatase levels, and elevated or normal serum FGF-23 levels. These tumours, usually small, benign, slow growing and difficult to discover, are mainly localized in soft tissues of the limbs. Histologically, phosphaturic mesenchymal tumours of the mixed connective tissue type are most common. Various imaging techniques have been suggested with variable results. Treatment of choice is total surgical resection of the tumour. Medical treatment includes oral phosphorus and calcitriol supplements, octreotide, cinacalcet, and monoclonal antibodies.
Los síndromes paraneoplásicos endocrinos constituyen manifestaciones a distancia de algunas neoplasias. Una forma infrecuente, pero cada vez más descrita, es la osteomalacia tumoral (OT), un trastorno hipofosfatémico secundario a la pérdida renal de fosfatos inducida por la secreción tumoral del factor de crecimiento fibroblástico 23 (FGF-23). Sus principales manifestaciones bioquímicas son la hipofosfatemia, la reabsorción tubular de fosfatos inadecuadamente normal o baja, los niveles bajos de calcitriol, la fosfatasa alcalina elevada y el FGF-23 sérico elevado o normal. Los tumores asociados a la OT suelen ser pequeños, benignos, de lento crecimiento, de difícil localización y con predominio en las partes blandas de los miembros. La histología más frecuente son los tumores mesenquimales fosfatúricos tipo tejido conectivo mixto. Se han propuesto varias técnicas de imagen para su identificación con resultados variables. El tratamiento de elección es la resección quirúrgica completa de la lesión. Otras alternativas terapéuticas son las sales de fósforo, el calcitriol, la octreótida, el cinacalcet y los anticuerpos monoclonales.
Paraneoplastic syndromes include a constellation of signs and symptoms occurring as a consequence of the distant effects of a tumour on different organs and systems. Such effects may be mediated by molecules with hormonal action, growth factors, cytokines, the development of autoimmunity, and other unknown factors. The term ‘ectopic’ refers to the secretion of a hormone by tissues which physiologically do not exert such a function. However, hormones secreted by tumours are generally present in non-malignant precursor cells, usually in small amounts. Thus, most endocrine manifestations of tumours are caused by eutopic hormone secretion by cells previously programmed to secrete hormones.
Inappropriate hormone secretion in neoplasms is characterized by its being rarely suppressible. The generation of abnormal or incompletely processed molecules with limited biological activity and, occasionally, peptides related to certain hormones (e.g. insulin-like growth factor type II (IGF-II), parathyroid hormone-related peptide) is also common. Table 1 lists the main hormones involved in paraneoplastic syndromes.
Main molecules involved in endocrine paraneoplastic syndromes.
| Factors for hypercalcemia |
| Parathyroid hormone-related peptide (PTHrp) |
| 1-25-dihydroxycolecalciferol (calcitriol) |
| Tumour necrosis factor (TNF) |
| Prostaglandins |
| Parathyroid hormone (PTH) |
| Adrenocorticotropic hormone (ACTH) |
| Vasopressin |
| Human chorionic gonadotropin (HCG) |
| Erythropoietin |
| Calcitonin |
| Insulin-like growth factor type II (IGF-II) |
| Somatotropin-releasing hormone (GHRH) |
| Human placental lactogen (hPL) |
| Growth hormone (GH) |
| Corticotropin-releasing hormone (CRH) |
| Atrial natriuretic peptide (ANP) |
| Endothelin |
| Renin |
| Gastrointestinal hormones (somatostatin, gastrin-releasing peptide, etc.) |
| Fibroblast growth factor 23 (FGF-23) |
Osteomalacia is a metabolic bone disease characterized by a defect in bone matrix mineralization. This disorder is called rickets in childhood, when growth cartilage is also altered. The mineralization process requires adequate calcium and phosphate levels, and preserved cell function and bone matrix structure. Thus, the two main causes of osteomalacia are impaired vitamin D and phosphate metabolism. There are other uncommon conditions that may interfere with bone mineralization, including changes in alkaline phosphatase, some drugs, and bone matrix disorders (Table 2). Among hypophosphatemic osteomalacias, an uncommon aetiology is tumour-induced osteomalacia [TIO]), also called oncogenic osteomalacia.1,2 TIO is a paraneoplastic syndrome caused by renal loss of phosphorus. It was first described by McCance in 1947,3 although its link to a humoral factor is attributed to Prader in 1959. The term phosphatonins was later used to refer to phosphaturic humoral factors, and at the beginning of this century, the central role of fibroblast growth factor 23 (FGF-23) in hypophosphatemic osteomalacia was identified.4 Less than 400 cases have been reported in the medical literature, which reflects its low incidence, its difficult identification and, probably, its underdiagnosis.5 TIO may occur at any age, but is more common in adults aged 50–70years.6
Causes of osteomalacia.
| Vitamin D deficiency or metabolic disorders |
| Inadequate intake |
| Low sunlight exposure |
| Use of creams with ultraviolet radiation filter (protection factor >8) |
| Skin hyperpigmentation |
| Advanced age (decreased skin synthesis of vitamin D) |
| Malabsorption (gastrectomy, gastric bypass, celiac disease, cholestyramine, chronic cholestasis, etc.) |
| Increased vitamin D catabolism (anticonvulsants, tuberculostatics, corticosteroids, etc.) |
| Defective hepatic 25-hydroxylation (chronic severe liver disease) |
| Defective renal 1α-hydroxylation (chronic renal failure, vitamin-D dependent rickets type I) |
| Renal loss of 25-hydroxyvitamin D (nephrotic syndrome) |
| 1-25(OH)2D receptor abnormalities (chronic renal failure, vitamin-D dependent rickets type II)) |
| Hypophosphatemic osteomalacia |
| X-linked hypophosphatemic rickets |
| Autosomal hypophosphatemic rickets (dominant, recessive) |
| Hypophosphatemic rickets with hypercalciuria |
| Bone fibrous dysplasia |
| Fanconi syndrome (congenital, acquired) |
| Acquired tubulopathies (renal tubular acidosis, heavy metals, drugs, multiple myeloma, etc.) |
| Intravenous iron therapy |
| Antiretroviral therapy |
| Low phosphate intake associated with the use of nonabsorbable antacids |
| Tumour-induced osteomalacia |
| Other causes of osteomalacia |
| Ureterosigmoidostomy |
| Drug treatment with fluorinate compounds, bisphosphonates, aluminium |
| Congenital hypophosphatasia |
| Fibrogenesis imperfecta |
| Axial osteomalacia |
Chronic variants of hypophosphatemia are associated with clinical muscular (myalgia, weakness, proximal myopathy) and bone signs (rickets in children and osteomalacia in adults). Three main pathophysiological mechanisms have been reported: redistribution (from the internal environment to the inside of cells), decreased intestinal absorption, and increased renal excretion of phosphorus. The main regulators of phosphorus metabolism are parathyroid hormone (PTH), 1-25 dihydroxyvitamin D or calcitriol (1-25[OH]2D), and FGF-23. FGF-23 is normally expressed by osteocytes and regulates phosphorus metabolism and vitamin D through binding to the Klotho-FGF receptor complex.7 In the kidney, it acts by decreasing tubular reabsorption of phosphate through the inhibition of expression of type 2a and 2c sodium/phosphate cotransporters (NaPi-2a 2c) and by inhibiting renal 1α-hydroxylase activity, thus causing hypophosphatemia, hyperphosphaturia, and low calcitriol levels. FGF-23 is involved in the pathophysiology of several hypophosphatemic disorders. Other phosphaturic disorders identified include fibroblastic growth factor 7 (FGF-7), matrix extracellular phosphoglycoprotein (MEPE), and secreted frizzled-related protein 4 (sFRP4), but their role in TIO development has yet to be defined.
Clinical manifestationsThe symptoms and signs of TIO are similar to those of familial hypophosphatemic osteomalacia. The main clinical manifestations in adults include bone pain, proximal muscle weakness, and insufficiency fractures, especially in weight-bearing bones such as the pelvis and lower limbs. Radiologically, multiple pseudofractures or Looser-Milkman zones, a characteristic but non-specific sign, are seen as radiolucent bands perpendicular to the cortex, usually bilateral and symmetrical, which sometimes progress to complete fractures. They are most commonly located in the ribs, pubic rami, external margin of scapula, internal margin of the proximal femur, and metatarsal bones. Bone scintigraphy, more sensitive than X-rays for the location of pseudofractures, shows isolated hyperuptake areas that may be confused with bone metastases. A generalized increase in isotopic uptake (superscan image) may also be seen, especially in the cranium, jaw, and chondrocostal joints, due to secondary hyperparathyroidism. The clinical picture of TIO is insidious, progressive and with non-specific symptoms, and it is therefore usually confused with rheumatic, oncological, psychiatric, and other disease. This results in a variable delay in correct diagnosis, which may be up to 20 years. In addition, the delay reported between the biochemical diagnosis of hypophosphatemic osteomalacia and tumour identification ranges from 2 to 5 years, because the vast majority of tumours are benign but small, occult or poorly evident, and difficult to locate.5
Biochemical findingsBiochemical diagnosis is based on the finding of hypophosphatemia, hyperphosphaturia, the decreased tubular reabsorption of phosphate (TRP), low or inappropriately normal serum 1-25(OH)2D levels, and high or inappropriately normal plasma FGF-23 levels. Under normal physiological conditions, 85%–95% of phosphorus filtered by renal glomerulus is mostly reabsorbed in the proximal tubule (∼85%) and, to a lesser extent, in the distal convoluted tubule (∼15%). The normal range of phosphaturia is wide, but in the presence of hypophosphatemia, a TRP lower than 95% suggests inappropriate urinary loss. Some authors recommend assessment of the ratio of the tubular maximum reabsorption of phosphate to the glomerular filtration rate (TmP/GFR), as its values are independent of plasma phosphorus level and kidney function, although it varies depending on age and sex and has not been validated in large populations.8 Secondary hyperparathyroidism may occur as a physiological response to low 1-25(OH)2D levels. Total and bone alkaline phosphatase are variably increased due to increased osteoblastic activity. The measurement of serum FGF-23 levels using an enzyme-linked immunosorbent assay (ELISA) may support the clinical diagnosis with an estimated sensitivity ranging from 23% to 86% in TIO with no tumour identified and from 38% to 100% in TIO with documented tumour; this variability may be attributed to the heterogeneity of the patient groups studied.9,10 However, this procedure is not always available, and TIOs with normal serum FGF-23 levels have been reported.11 A differential diagnosis includes related genetic disorders (Table 2), particularly some forms of autosomal recessive familial hypophosphatemic rickets with late onset in adults; hereditary or acquired Fanconi syndrome, other forms of tubulopathies with renal phosphate loss (heavy metals, drugs, multiple myeloma, etc.), and various disorders associated with hypophosphatemia (liver failure, malabsorption, alcoholism, etc.) should also be taken into consideration in a differential diagnosis.
HistopathologyTumours causing TIO are usually small, benign, and slow-growing, and are often located in the limbs, both in bone and soft tissue. They have also been found in the paranasal sinuses, nasopharynx, brain, ovaries, spine, and pelvis.12–14 In some cases the tumour is not localized. In one series reported, this occurred in more than one third of patients.5 Some authors refer to this condition as a TIO-like syndrome or secondary TIO, which has been associated with various tumours such as sarcoma, prostate and breast carcinoma, multiple myeloma, chronic lymphocytic leukaemia, and small-cell carcinoma. The paraneoplastic syndrome currently recognized as TIO is associated with tumours of mesenchymal origin, usually benign, previously described as giant-cell tumours, ossifying fibromas, osteoblastomas, granulomas, hemangiopericytomas, and others. Weidner et al. proposed the term phosphaturic mesenchymal tumours, and their division into the following categories: mixed connective tissue, osteoblastoma-like tumours, non-ossifying fibroma-like tumours, ossifying fibroma-like tumours, and metastatic tumours.15 Overall, they represent a distinct histopathological, although morphologically heterogeneous, entity,16 with the first variant being the most common and accounting for approximately 75% of all cases. In the literature on the subject in English they are identified as phosphaturic mesenchymal tumour-mixed connective tissue variants (PMT-MCT). They are characterized by having giant cells similar to osteoclasts, myxoid or chondromyxoid stroma, low or no mitotic activity, ossified areas, and significant vascularization, with vessels of different sizes and morphological patterns. The previous lack of uniform criteria for their recognition could be responsible for a certain confusion in diagnosis, although as a last resort, a bone biopsy without prior bone decalcification marked with tetracycline may be performed to show increased osteoid and a mineralization lag time longer than 100 days.17 Although tumours are usually benign, malignant and metastatic presentations have also been reported.18
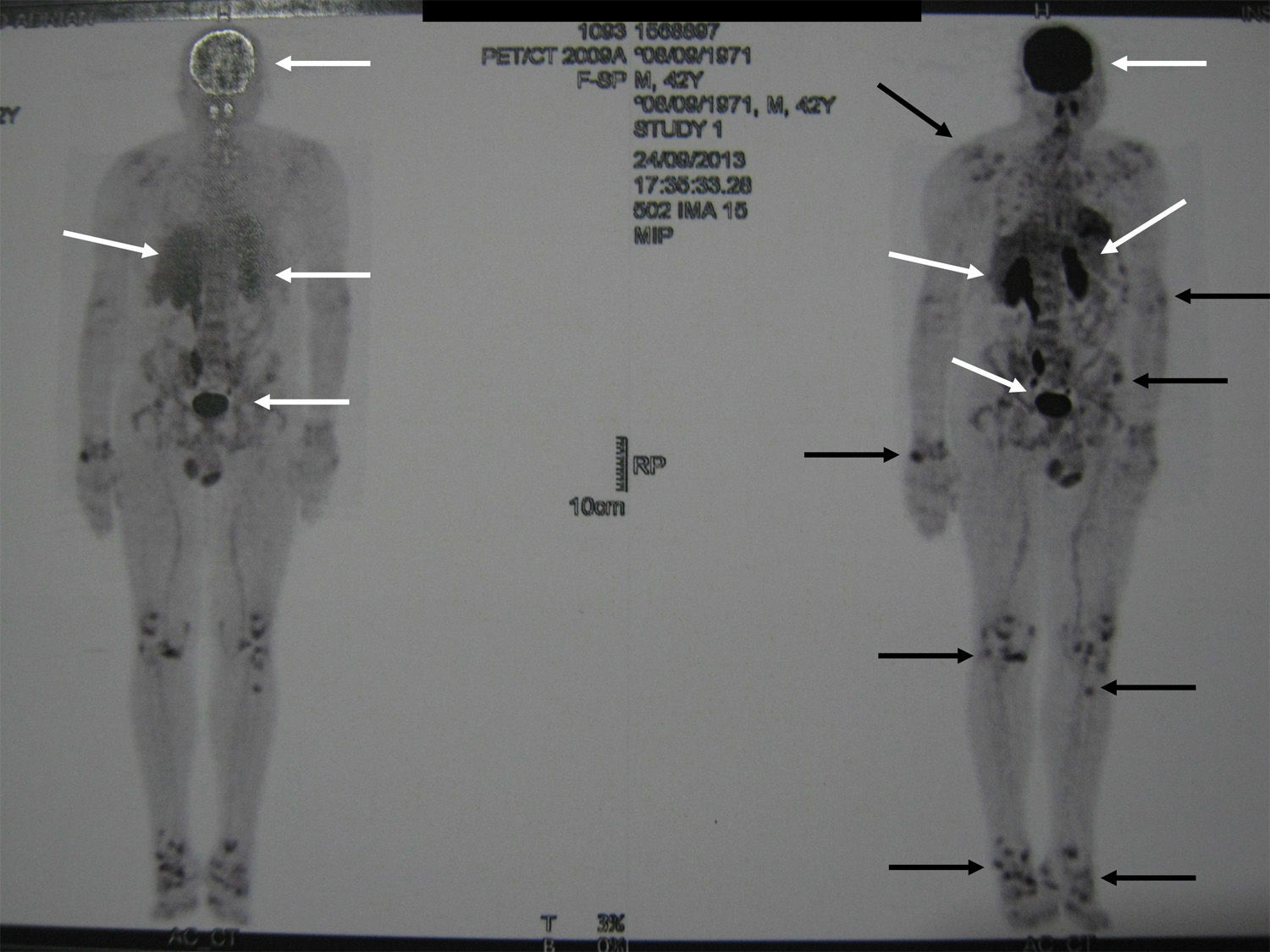
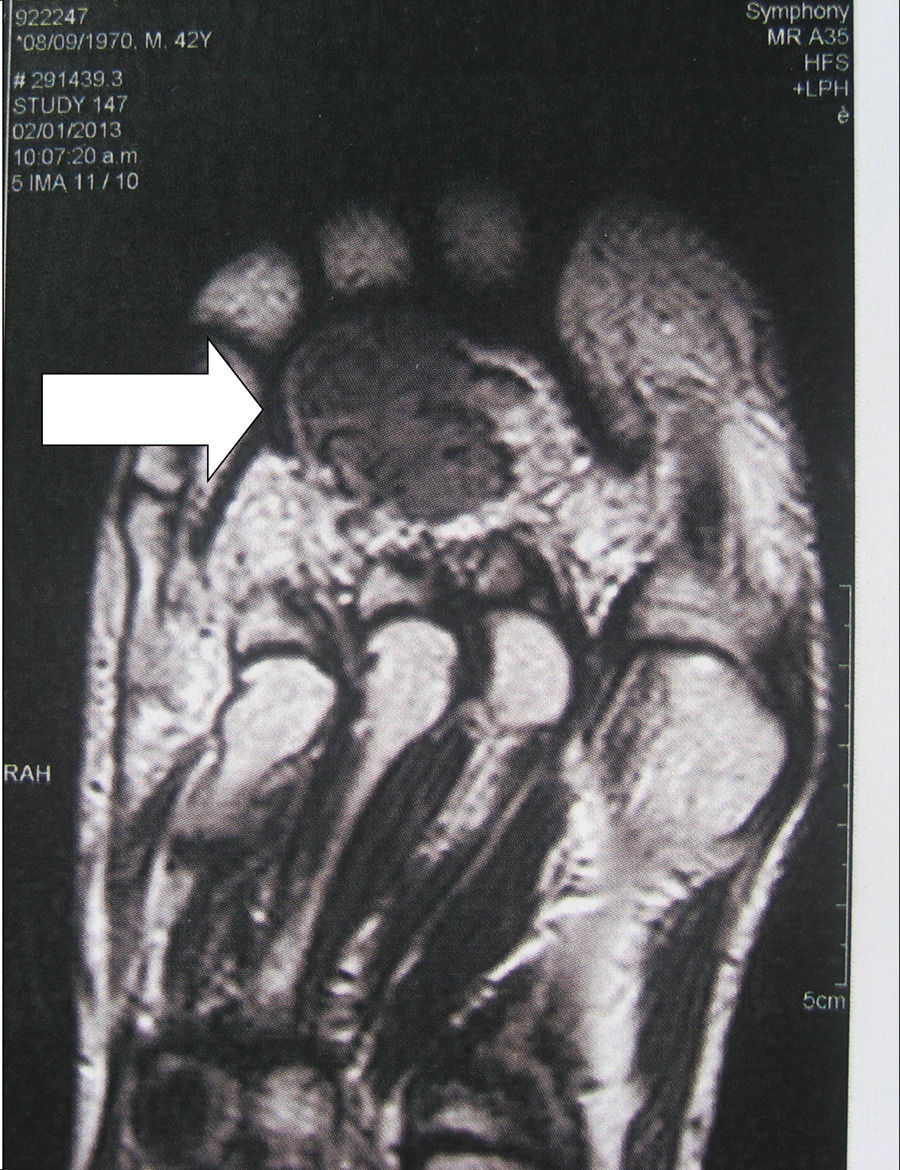
Localization methodsA comprehensive search should be performed by means of a complete physical examination. When the tumour is not evident, imaging techniques and functional studies are used. Because of the characteristics of these tumours, conventional imaging techniques are often unable to detect them. The localization methods proposed include a computed tomography (CT) scan of the paranasal sinuses, whole body magnetic resonance imaging (MRI),19 positron emission tomography with fluorodeoxyglucose (FDG-PET/CT),20 scintigraphy with octreotide labelled with 111In (octreoscan),21 and scintigraphy with 99Tc or 201Th-sestamibi.22 In recent years, Ga68-DOTANOC PET/CT (using a modified octreotide molecule),23 or venous sampling with FGF-23 measurement in areas where functional studies suggest suspect lesions, have been incorporated in recent years.22 FDG-PET/CT is a highly-sensitive method, but has a low specificity, particularly in patients with many areas of pseudofracture, healing fractures, or lytic areas20 (Fig. 1). Physiological uptake in brain, liver, and spleen may make tumour identification in those areas difficult, and other imaging methods are recommended in order that such locations can be assessed. Whole body scans should be performed, because distal portions of the head and lower limbs, where many of these tumours are located, are sometimes excluded. The identification of suspect hypermetabolic foci requires anatomical confirmation with MRI or CT (Fig. 2). Despite advances in the above procedures, the tumour is not always located, in which case repeated imaging studies are recommended one or two years later.2
. Note the physiological uptake in brain, liver, spleen, and urinary tract (white arrows).")

The treatment of choice is complete surgical resection of the tumour with a wide margin, because postoperative recurrence has been reported.24 When surgery is successful, the clinical and biochemical picture progressively resolves, although some manifestations may persist for several months and there are usually permanent bone sequelae. Phosphate levels commonly normalize within 2–10 days of surgery. Late recurrence due to metastasis is possible, but uncommon. If this occurs, lung involvement has most commonly been reported.5 Until surgery is performed, and in patients in whom complete surgical resection is not possible or postoperative recurrence occurs, medical treatment should be administered with oral supplements of phosphorus salts (15–60mg/kg/day, usually 1–3g/day of elemental phosphorus in 4–6divided daily doses) and calcitriol (0.50–1.0μg/day) taken separately, which may achieve clinical and biochemical improvement. Treatment should be individualized based on age, weight, PTH levels, and kidney function. This therapy should be closely monitored so that the biochemical response may be adjusted according to tolerability and to prevent hypercalcemia, hypercalciuria, nephrolithiasis, and nephrocalcinosis. Somatostatin receptor agonists (parenteral long-acting octreotide) have been tried with variable responses.25 Cinacalcet, an agonist of the calcium-sensing receptor, is another potential treatment based on interactions between FGF-23 and PTH.26 There have also been reports of patients treated by radiofrequency ablation and intratumoral ethanol injection, with promising results.27 The use of combinations of chemotherapy and radiotherapy has given limited results. A novel approach is the use of monoclonal antibodies that interrupt the interaction of FGF-23 with its receptor.28
ConclusionThe cases reported in the literature exemplify the main characteristics of TIO: delayed diagnosis, difficult localization of tumours (usually benign in nature), predominant occurrence in the lower limbs, cure after complete resection, and potential recurrence.5,29–31 The majority of cases worldwide have been reported in recent years, suggesting that the condition was probably underdiagnosed previously. It is thus important to disseminate both understanding of TIO and uniform criteria for its identification. The World Health Organization estimates that there are approximately 7000 rare, uncommon, or minority diseases affecting 7% of the population worldwide. This concept encompasses a number of diseases with a low prevalence, diagnostic difficulties, lack of information and scientific knowledge, limitations in research due to lack of investment or resources, and deficient therapies. TIO is an uncommon disease and represents a true challenge for internists. Despite this, and bearing in mind the chances of curing the disease with complete tumour resection, it is important to take the condition into account and to request and evaluate adequate diagnostic tests to identify and resect the phosphaturic tumour as soon as possible.
FundingThe authors have received no grants or funding for the conduct of this study.
Authorship/collaboratorsGuillermo Alonso and Mariela Varsavsky participated in the study conception, study design, data collection, data analysis and interpretation, and the writing, review and approval of the manuscript submitted.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Alonso G, Varsavsky M. Osteomalacia tumoral: un síndrome paraneoplásico emergente. Endocrinol Nutr. 2016;63:181–186.
