


Biochemical tests related to calcium and phosphorus metabolism have traditionally been considered as a reliable tool to differentiate familial hypocalciuric hypercalcemia (FHH) from primary hyperparathyroidism (PHPT). However, diagnosis may sometimes be difficult even for experienced clinicians. Our objective was to assess the accuracy of diagnostic tests in FHH and the circumstances in which genetic studies are required.
Patients and methodsA descriptive study was conducted of two families with hypercalcemia and suspected atypical FHH. Urinary calcium excretion was measured in 24-h urine using different tests (calcium excretion (CE), urinary calcium/creatinine clearance ratio (UCCR)), and serum PTH and 25-hydroxyvitamin D levels were tested. Index cases underwent genetic study.
ResultsOne patient from the first family showed overt, persistent hypercalciuria with values more consistent with PHPT than with FHH if we consider, as proposed by guidelines, a UCCR lower than 0.01 as diagnostic of FHH and a value higher than 0.02 as diagnostic of PHPT. The index case of the second family underwent surgery for a parathyroid adenoma. Both cases had a mutation c. 164C>T (Pro55Leu) in exon 2 in heterozygosis.
ConclusionsAccording to current clinical guidelines, definitive diagnosis of FHH requires genetic confirmation, which allowed in our case for detection of two families with FHH and atypical clinical presentations. We think that rational use of genetic tests may avoid unnecessary surgery and excess monitoring costs.
Tradicionalmente se ha pensado que las pruebas bioquímicas del metabolismo fosfocálcico permiten diferenciar el hiperparatiroidismo primario (HPT1) y la hipercalcemia hipocalciúrica familiar (HHF) pero hay casos de difícil diagnóstico incluso para clínicos experimentados.
Nos planteamos como objetivo evaluar la validez de las pruebas diagnósticas de la HHF así como la correcta indicación del estudio genético.
Pacientes y métodosHemos realizado un estudio descriptivo de 2 familias con hipercalcemia y sospecha de HHF de características atípicas.
En orina de 24 h hemos valorado los índices de excreción urinaria de calcio (eliminación de calcio [CE], cociente calcio/creatinina [CR] y cociente aclaramiento de calcio/aclaramiento de creatinina [CCCR]), junto con las concentraciones séricas de PTH y 25 hidroxivitamina D. A los casos índices se les realizó el estudio genético.
ResultadosUna paciente presentó hipercalciuria franca y persistente con valores más concordantes con HPT1 que de HHF si consideramos, como proponen las guías, un CCCR inferior a 0,01 como indicativo de HHF y superior a 0,02 como HPT1. Al caso índice de la segunda familia se le extirpó un adenoma de paratiroides. En ambos casos índice, encontramos la misma mutación c. 164C>T (p.Pro55Leu) en el exón 2 en heterocigosis descrita como responsable de HHF.
ConclusionesEl diagnóstico definitivo de HHF en las guías clínicas actuales requiere confirmación genética, lo cual ha permitido en nuestro caso la detección de 2 familias con HHF y características clínicas atípicas. En nuestra opinión el uso racional de estas pruebas ante la sospecha de HFF puede evitar intervenciones quirúrgicas innecesarias y gastos excesivos en su monitorización.
Familial hypercalcemia is not uncommon in endocrinology offices. It is a hereditary disorder with an autosomal dominant transmission in most cases1,2 covering a spectrum of diseases including multiple endocrine neoplasia type 1 and 2 (MEN 1 and 2), hyperparathyroidism associated to jaw tumor, isolated familial hyperparathyroidism, and familial hypocalciuric hypercalcemia (FHH). Differentiation of the five syndromes is sometimes difficult, but has profound implications for the patient and his/her family.
Availability of specific genetic tests has improved diagnostic accuracy and simplified monitoring, but these tests are costly and often not easily accessible, and therefore require a rational use.
Inactivating mutations in the calcium-sensing receptor gene (CaSR), located in the long arm of chromosome 3, may affect a single allele (heterozygosis), which results in the phenotype characteristic of FHH, or both alleles (homozygosis or compound heterozygosis if no consanguinity exists), which leads to severe neonatal hyperparathyroidism, so that the grade of gene defect accounts for the great disparity in phenotypic presentation.3
Some 200 different mutations4 have been reported in FHH. The end result of such mutations is a decreased number of functional receptors in parathyroid cells and kidney, which makes them partially resistant to calcium, so that high calcium levels are required to decrease PTH and calcium and magnesium are reabsorbed in the kidney even in the presence of hypercalcemia. FHH therefore causes persistent hypercalcemia, hypocalciuria which is either absolute (typically less than 200mg/day) or relative to plasma calcium (urinary calcium/creatinine clearance ratio [UCCR] less than 0.01), and normal PTH levels, although 20% of patients have elevated PTH levels.5,6 FHH is a disorder which is considered to be benign and asymptomatic and does not appear to shorten life expectancy, but chondrocalcinosis has been reported in some adults and may confer an increased susceptibility to pancreatitis.7 It is important to distinguish this condition from primary hyperparathyroidism (PHPT), which is usually easy in the typical cases of both conditions because surgery is unnecessary and ineffective in FHH.
The clinical spectrum of mutations in the calcium-sensing receptor is being widened with the genetic study, and hypercalciuria, stones8 and, exceptionally, parathyroid adenoma have been reported in patients with FHH.9
Patients and methodsThis was a descriptive study of two families from the same geographic area but with no common known family history which had shown hypercalcemia during years of follow-up and in which a genetic study had recently confirmed the presence of FHH, despite their discordant clinical and biochemical data.
The index cases of the first family, with four affected members, there was a 35-year-old woman 54.4kg in weight and 158cm in height and with a body mass index (BMI) of 21kg/m2 in whom laboratory tests performed for a headache detected increased calcium levels (11.1mg/dL), slightly decreased phosphorus levels (2.4mg/dL), and normal magnesium and creatinine levels. Other test results included 24-h urinary calcium of 272mg, tubular phosphate reabsorption (TPR) of 70%, urinary calcium/creatinine clearance ratio (UCCR) of 0.0137, elevated intact PTH levels (110pg/mL), and 25-hydroxyvitamin D deficiency (24.8nmol/L). parathyroid ultrasonography and a Tc-sestamibi scan showed no enlargement of parathyroid glands, adenoma, or ectopic tumors. Abdominal ultrasonography was normal, as was bone densitometry of the spine, showing a T score of −0.02 and a Z score of 0.50.
Family study revealed asymptomatic hypercalcemia in the 70-year-old father of the woman and in her two sons, aged 18 and 12 years. Soon after diagnosis, the eldest son experienced repeated episodes of renal colic and gross hematuria but passed no stones, which were not visible in standard abdominal X-rays. He attended the emergency room of our hospital for that reason in 2004, 2008, and 2010.
In the second family, the index case was a 39-year-old male 92kg in weight and 173cm in height (BMI, 30.7kg/m2) in whom hypercalcemia was discovered during work-up for hyperlipidemia. Further investigation revealed hypercalcemia in two of four brothers, including the index case, and also in the mother and one uncle. Initial laboratory tests showed 11.7mg/dL of calcium, 2.74mg/dL of phosphorus, and 2.19mg/dL of magnesium. Urinary calcium levels were 82mg/24h, UCCR 0.003, urinary phosphate levels 1.254mg/24h, and TPR, 80%. Intact PTH was elevated (72pg/mL) and 25-hydroxyvitamin D levels were decreased (40nmol/L [normal range, 50–250]). Bone densitometry showed spine osteopenia (T score −1.30 and Z score −1.20). Thyroid ultrasonography was normal, but a Tc-sestamibi scan showed pathological retention of the radiotracer in the lower portion of left thyroid lobe consistent with a parathyroid adenoma in the left inferior parathyroid gland.
Calcium phosphorus, magnesium, alkaline phosphatase, and creatinine were measured in fasting conditions using a Cobas c711 Hitachi analyzer (Roche Diagnostics).
Calcium and phosphate levels and TPR were tested in 24-h urine while patients were taking an unrestricted diet, and diuretic use by patients was ruled out. The three most commonly used indices of urinary calcium excretion were assessed, defining hypercalciuria as calcium excretion (CE) greater than 4mg/kg/24h or 240mg in females and 300mg in males or a calcium/creatinine ratio (CR) greater than 0.2mg/kg. A UCCR (calcium in 24-h urine×serum creatinine/serum calcium×urinary creatinine) less than 0.01 was considered indicative of hypocalciuria.
Chemoluminescence (Inmulite 2000, Siemens) was used to measure intact PTH (normal range, 11–67pg/mL) and 25-hydroxyvitamin D (normal range, 50–250nmol/L) levels. Vitamin levels less than 25nmol/L were considered indicative of severe deficiency, while levels ranging from 25 to 50nmol/L and from 50 to 75nmol/L were, respectively defined as insufficient and suboptimal in Andalusia10).
Index cases of each family were requested to consent for a molecular genetic study for the calcium receptor gene, which was performed at the department of biochemistry and molecular genetics of Hospital Clínico in Barcelona.
DNA was extracted from white blood cells from whole blood using QIAmp DNA (QIAGEN). For CaSR gene analysis (Ensembl: ENSG00000036828), coding regions of exons 2, 3, 4, 5, 6, and 7 were amplified by PCR using flanking primers (http://frodo.wi.mit.edu/primer3/ was used for primer design). PCR products were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and were purified using the Millipore system (96-well plates Multiscreen PCRu96 & Montage seq96). Products were then analyzed in an ABI Prism Genetic Analyzer 3130.xl (Applied Biosystems).
ResultsOf the three renal calcium excretion indices (CE, CR, and UCCR), UCCR is considered to be the index of choice because of its better discriminating power. Guidelines recommend for UCCR a cut-off point less than 0.01 for diagnosis of FHH, which means that more than 99% of filtered calcium is reabsorbed despite the presence of hypercalcemia, and a cut-off value higher than 0.02 for PHPT diagnosis.
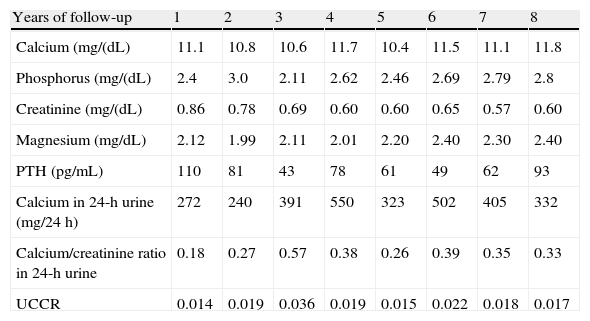
Table 1 shows the basic descriptive laboratory variables in the eight years of follow-up of the index case of the first family.
Basic descriptive laboratory variables in the index case of the first family.
| Years of follow-up | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Calcium (mg/(dL) | 11.1 | 10.8 | 10.6 | 11.7 | 10.4 | 11.5 | 11.1 | 11.8 |
| Phosphorus (mg/(dL) | 2.4 | 3.0 | 2.11 | 2.62 | 2.46 | 2.69 | 2.79 | 2.8 |
| Creatinine (mg/(dL) | 0.86 | 0.78 | 0.69 | 0.60 | 0.60 | 0.65 | 0.57 | 0.60 |
| Magnesium (mg/dL) | 2.12 | 1.99 | 2.11 | 2.01 | 2.20 | 2.40 | 2.30 | 2.40 |
| PTH (pg/mL) | 110 | 81 | 43 | 78 | 61 | 49 | 62 | 93 |
| Calcium in 24-h urine (mg/24h) | 272 | 240 | 391 | 550 | 323 | 502 | 405 | 332 |
| Calcium/creatinine ratio in 24-h urine | 0.18 | 0.27 | 0.57 | 0.38 | 0.26 | 0.39 | 0.35 | 0.33 |
| UCCR | 0.014 | 0.019 | 0.036 | 0.019 | 0.015 | 0.022 | 0.018 | 0.017 |
UCCR: urinary calcium/creatinine clearance ratio.
As shown by 24-h calcium elimination, our patient had frank, persistent hypercalciuria over time if CE and CR are considered. Assessment of UCCR results revealed intermediate values, and some clearly suggesting PHPT rather than FHH.
These high urinary calcium levels also coincide with an associated deficiency of vitamin D, whose concentration modulates the severity of the condition by causing an additional decrease in urinary calcium and often justifying the increased PTH levels seen in FHH and found in our patient.
Unlike her, her two sons have shown hypocalciuric hypocalcemia since 1996, with UCCR below or very slightly above 0.01. The youngest son has remained symptom-free and with normal 25-hydroxyvitamin D levels (80nmol/L). The eldest son experienced in 2005 three episodes of renal colic requiring treatment at the emergency room. He had at the time a plasma calcium level of 11.7mg/dL, a phosphorus level of 3.17mg/dL, 24-h urine CE of 248mg, UCCR of 0.0123, an inappropriately elevated PTH level of 84pg/mL, and a decreased 25-hydroxyvitamin D value of 49nmol/L. Abdominal and cervical ultrasonography and Tc-sestamibi scan were normal. Exploratory surgery with subtotal parathyroidectomy was performed, but no pathological changes were found in parathyroid glands, and hypercalcemia persisted after surgery. Oxalate and citrate levels in 24-h urine were normal, but uric acid excretion was persistently elevated (up to 1542mg/24h), which may suggest the presence of uric stones, but patient has not passed to date stones amenable to analysis.
Because of the discordant data in this family, genetic study of the index case was performed in 2010, revealing the c. 164C>T (Pro55Leu) mutation in exon 2, which has been reported to be responsible for FHH.
In the index case of the second family, patient age, calcium concentrations, osteopenia, and scan uptake led to also perform exploratory surgery in the neck, with hypocalciuria being explained by his low vitamin D levels. An adenoma in the left inferior parathyroid gland was resected at surgery, and PTH levels decreased from 72pg/mL during surgery to 47pg/mL after the procedure. Although total calcium level normalized the day after surgery to 9.8mg/dL, patient continued to show hypocalciuric hypercalcemia (UCCR less than 0.01 at all times) at subsequent annual follow-up visits to our clinic despite normalization of vitamin D levels after treatment. Results at the last follow-up included: calcium 11.5mg/dL, phosphorus 1.96mg/dL, magnesium 2.3mg/dL, UCCR 0.009, 25-hydroxyvitamin D 120nmol/L, and PTH 34.8pg/mL.
All other three members of the family remained symptom-free and had UCCR values also lower than 0.01 during the years of follow-up.
Based on discordant history data, surgery for parathyroid adenoma, and family data of hypocalciuric hypercalcemia, a genetic study was recently requested and also showed the c. 164C>T (Pro55Leu) mutation in exon 2 in heterozygosis reported to be responsible for FHH.
DiscussionIn recent years, availability of genetic tests has allowed for finding families with heterozygous mutations in the CaSR gene which show clinical features characteristic of hyperparathyroidism such as hypercalciuria, kidney stones and, more rarely, parathyroid adenoma. We monitored for years two families, with eight affected members, with suspected FHH. This was recently confirmed by genetic testing, but two of the patients were found to have a discordant course, consisting of sustained hypercalciuric hypercalcemia in one of them and parathyroid adenoma in the other.
Familial hypercalcemia follows an autosomal dominant pattern of inheritance, and is therefore diagnosed in families with several affected members, many of them young. When patients have asymptomatic hypercalcemia with relative hypocalciuria, especially if there is a history of ineffective parathyroid surgery, urinary and plasma levels should be investigated in several generations of relatives even if they are apparently unaffected. Calcium measurement in blood and urine from three members of a family gives less false positive results than a genetic study in an index case. If an individual subject has no affected relatives, FHH is highly unlikely,11 but cannot be ruled out because families with recessive inheritance have been reported,11 or de novo mutation may occur. No CaSR mutations are found in up to 30% of families, which may be due in some cases to presence of mutations in other regions (promoter, introns, etc.) or implication of other different genes.
The fact that genetic tests are not indispensable because of the benign nature of the condition, their cost, their high false negative rate, and their difficult accessibility for clinicians until recently have limited their routine use in FHH. They may, however, prevent unnecessary surgery and substantial expenses in doubtful cases.12
Most cases of FHH and familial PHPT do not usually pose problems in differential diagnosis, but both diseases have atypical presentations difficult to differentiate. Thus, normal PTH levels are found in 10% of PHPT, and high PTH levels are seen in 15–20% of patients with FHH. The guidelines12 propose cut-off points for UCCR of less than 0.01 for diagnosis of FHH and more than 0.02 for diagnosis of PHPT, which are widely quoted and accepted.13 A study14 recently assessed the discriminating power of the three renal indices and established an optimum cut-off point of 0.0115, but there may be overlapping in urinary calcium excretion between the two conditions.
A decreased renal calcium excretion, with a UCCR even less than 0.001, may be found in some patients with PHPT, especially if they also have vitamin D deficiency, which is currently so common in Spain and Andalusia as to be considered an epidemic,15–17 and patients with FHH may have hypercalciuria or, more rarely, stones,1,8 which causes diagnostic errors even in the hands of experienced clinicians.
Twenty percent of cases of FHH and up to 12% of those of PHPT would be inadequately classified and potentially mistreated, even if the cut-off point for UCCR is increased from less than 0.01 to 0.0115. A two-step diagnostic approach has therefore been suggested13 for doubtful cases. UCCR is first determined using a cut-off value of less than 0.020. This excludes two thirds of PHPTs, which will have a higher value, and will include 98% of FHHs. The next step would be to determine the CaSR gene in families with UCCR less than 0.020, which separates patients with mutations (FHH) from those with mutations (PHPT).
A potential cause of error is not assessing concentrations of vitamin D, which is indispensable in these patients because its deficiency causes decreased intestinal absorption and renal excretion of calcium, increased PTH levels, and inactivation of the calcium-sensing receptor.18 Patients with PHPT appear to have lower 25-hydroxyvitamin D levels as compared to controls, and renal calcium excretion in these patients positively correlates to these levels, reaching in patients with deficiency levels typical of FHH.10,19
On the other hand, it should be noted that intensity of the CaSR gene mutation is variable and conditions the extent of PTH secretion, tubular calcium reabsorption, and calcemia in an individual patient.
Our two index cases from both families with FHH had decreased levels of 25-hydroxyvitamin D. In one of them, vitamin D replacement therapy decreased PTH levels but, as reported in patients with FHH,20 did not change renal calcium excretion, which continued to be very low (UCCR less than 0.01). Absence of a sustained increase in PTH in patients with FHH contributes to its usually benign course and to the absence of bone involvement.21
One of the patients had sustained hypercalciuria, despite the associated vitamin D deficiency, with UCCR values higher than 0.02 for years of follow-up, which is exceptional. One of her sons had repeated renal colic associated to hypocalciuria and high uric acid levels in urine.
At least 9% of cases with persistent hypercalcemia following ineffective surgery22 are patients not diagnosed with FHH. Pathological examination of parathyroid glands showed a slight enlargement or mild hyperplasia, but no nodularity is usually found. We report a genetically proven case of FHH in which a TC-sestamibi scan revealed a left inferior parathyroid adenoma which was confirmed at surgery. This technique has an 85–100% sensitivity for localizing adenoma before surgery, and a specificity close to 100% if there is no concurrent thyroid involvement, as occurred in our case.23
It is unknown whether FHH is a risk factor24 for subsequent development of PHPT. Expression of CaSR protein is often reduced in adenomas from patients with PHPT.24 One may therefore speculate that total or partial loss of receptor function may stimulate proliferation of parathyroid cells. Association of homozygous inactivating CaSR mutations to PHPT due to multiple parathyroid adenomas and severe persistent hypercalcemia after surgery has recently been reported, suggesting that loss of receptor function may occasionally induce PHPT in adults.25 Both diseases may rarely occur together. A search in Medline found that four cases of parathyroid adenoma in patients with genetically proven FHH have been reported in the past 10 years.8,9,26,27 This could be a simple coincidence, as PHPT is a common disease, but presence of parathyroid adenomas in different generations of one of our families does not appear to be due to chance.9
25-Hydroxyvitamin D deficiency in PHPT is associated to more aggressive evolution with greater bone involvement, and new guidelines28 therefore recommend vitamin D replacement.
For some authors, vitamin D deficiency may have a role in development of parathyroid adenoma, and they suggest that chronic deficiency accelerates adenoma growth9,19 and that vitamin D supplementation may reverse this process by acting upon the normal allele of FHH patients, but conflicting data are available.29,30
To sum up, availability of genetic studies is allowing for expanding the clinical spectrum of FHH. For current clinical guidelines,11 final diagnosis of FHH requires genetic testing. We analyzed the current genetic screening protocols in patients with suspected FHH for rational use. A genetic test for the CaSR mutation in atypical cases, such as the ones reported here, may prevent unnecessary surgery and, in our opinion, excess follow-up costs.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Fernández López I, et al. Utilidad del estudio genético en la hipercalcemia hipocalciúrica familiar en familias con presentaciones clínicas atípicas. Endocrinol Nutr. 2011;58:325–30.
