


El síndrome de McCune-Albright (SMA) es una condición heterogénea e infrecuente causada por la mutación postcigótica, somática y esporádica del gen GNAS que codifica la subunidad alfa estimuladora (αs) del receptor acoplado a proteína G1. Clínicamente, este síndrome consta de una tríada caracterizada por displasia fibrosa ósea, manchas café con leche y endocrinopatías hiperfuncionantes tales como pubertad precoz, hipertiroidismo, exceso de hormona de crecimiento (GH), hiperprolactinemia e hipercortisolismo; sin embargo, con al menos 2 de 3 características clínicas se hace el diagnóstico de SMA1,2.
Presentamos el caso de un paciente varón de 16 años de edad, natural y procedente de Mérida - Venezuela, sin antecedentes familiares de consanguinidad y con antecedentes personales de múltiples fracturas de fémur secundarias a displasia fibrosa poliostótica diagnosticada a los 3 años, quien consultó a la unidad de Endocrinología por presentar talla alta, refiriendo además cefalea holocraneana continua, de fuerte intensidad e hiposmia acentuada en los últimos años.
Al examen físico presentaba un peso de 93 kg (mayor del percentil 97), talla 183cm (mayor del percentil 97), con un potencial genético de talla (suma de la talla de ambos padres+12,5cm/2) de 169±10cm, índice de masa corporal (IMC) de 26,6 kg/m2 y presión arterial 120/70mmHg. Se evidenció manchas café con leche de 5,5 y 7cm de diámetro en la región posterior del cuello y en el glúteo derecho, respectivamente, ambas de bordes irregulares. Además, mostraba deformidad craneofacial caracterizada por macrocefalia, prominencia frontal e hipertelorismo, y presencia de tutores externos en ambos miembros inferiores debido a múltiples fracturas. Destacaba distribución androide del vello corporal, con testes de 25ml cada uno.
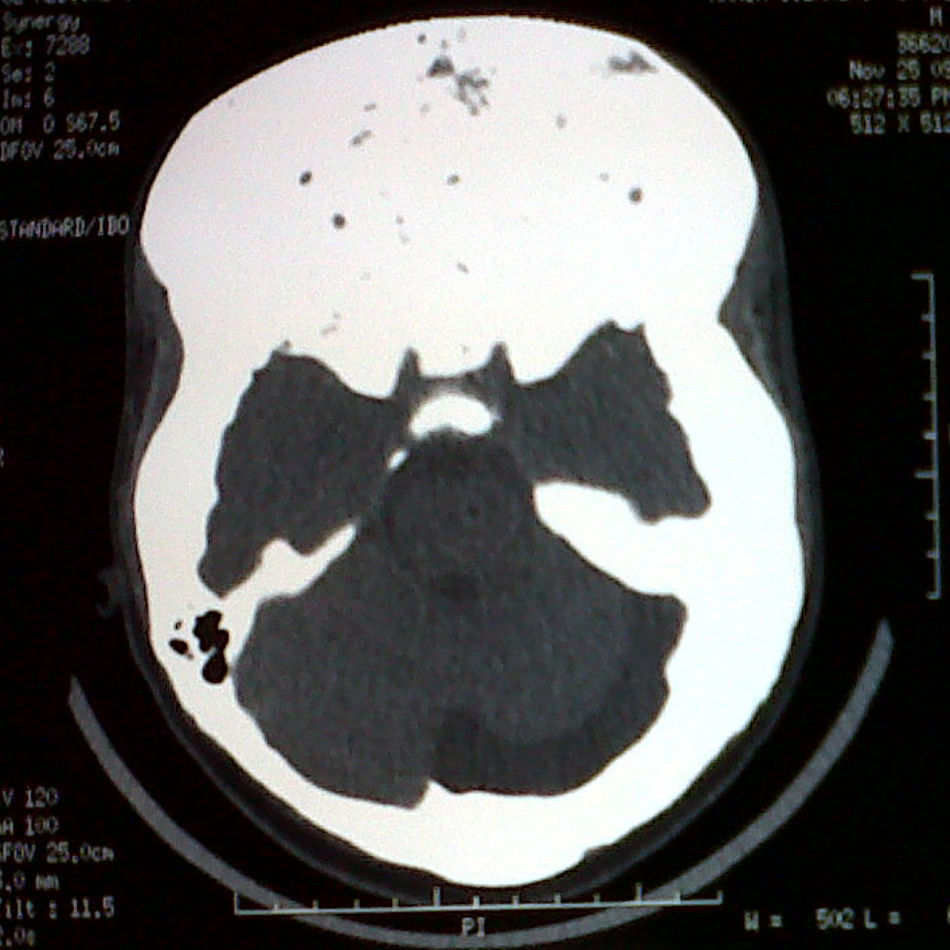
Se solicitaron análisis de laboratorio en los que se encontró una glucemia en ayunas normal, calcio: 8,8mg/dL (valor normal (VN):8,7-10,3), magnesio 2,1mg/dL (VN: 1,40-2,40), fósforo 2,2mg/dL (VN: 2,7-4,5mg/dL), hormona paratiroidea 35,1pg/mL (VN: 10-69), fosfatasa alcalina 1.396,1 UI/L (VN: 98-271), somatotropina (GH) basal 7,1μg/L (VN: 0-2,5), GH a las 2 horas postcarga con 75 gr de glucosa de 5,4μg/L (VN: menor de 1), IGF-1 (del inglés, insulin-like growth factor): 725ng/mL (VN: 72-385), tirotropina y tiroxina libre normales, cortisol basal 8,7μg/dL (VN:5-25), prolactina 57ng/mL (VN hombres: 0-15), hormona folículoestimulante 0,35 mUI/mL (VN: 0,7-11), hormona luteinizante 0,2 mUI/mL (VN: 0,8-7,6) y testosterona total: 92,8ng/dL (VN: 286-1.511). La radiografía de mano y muñeca izquierda reveló una edad ósea de 15 años. Además, se solicitó tomografía computarizada, observándose aumento de volumen de la masa ósea en bóveda craneana y estructuras de la región frontal, orbitaria y mastoidea compatibles con displasia fibrosa, y engrosamiento de los huesos de la región sellar lo cual no permitió una adecuada visualización de la hipófisis (fig. 1). No se pudo realizar resonancia magnética hipofisaria con gadolinio debido al uso de tutores externos y clavos endomedulares en miembros inferiores. Se indicó, por tanto, campimetría computarizada que mostró índices de confiabilidad y umbral foveal normal bilateral; sin embargo, en el campo visual del ojo derecho se observó una discreta disminución de la sensibilidad retiniana, mancha ciega aumentada de tamaño y escotoma focal temporal superior compatible con lesión prequiasmática de dicho ojo. Por su parte, el ojo izquierdo presentó una discreta disminución de sensibilidad retiniana, con mancha ciega de tamaño normal y sin escotomas.

En vista de los hallazgos clínicos, analíticos y pruebas de imagen se indicó octreotide LAR 20mg mensual, cabergolina 0,5mg 2 veces a la semana, undecanoato de testosterona 1.000mg trimestral, y ácido zoledrónico 5mg anual. El paciente se reevaluó al cabo de 3 meses, refiriendo mejoría leve de la cefalea y evidenciando un valor de GH basal de 2,4μg/L, GH a las 2 horas postcarga con 75 g de glucosa: 2,5μg/L, IGF-1 basal: 105ng/mL, prolactina: 37ng/mL, testosterona total: 1.290ng/dL y fosfatasa alcalina: 1.126,2 UI/L. Ante estos resultados se aumentó la dosis de octreotide LAR a 30mg mensual, manteniendo igual el resto de las indicaciones médicas.
Este paciente presentaba un SMA debido a la coexistencia de displasia fibrosa poliostótica, manchas café con leche y endocrinopatías como exceso de GH e hiperprolactinemia. La prevalencia de esta enfermedad se desconoce, pero se estima que sea de 1/100.000 a 1/1.000.000 de nacidos vivos, siendo más frecuente en el sexo femenino1. Por su parte, la displasia fibrosa puede ser monostótica cuando compromete un solo hueso o poliostótica cuando compromete más de 2 huesos, siendo la primera forma más frecuente y evidenciándose en el 7% de los tumores benignos óseos1,2.
Aproximadamente, el 20% de los pacientes que cursan con SMA presentan exceso de GH, observándose en los mismos una elevada prevalencia de hiperprolactinemia concomitante (71-92%). El 33% de ellos es debido a un microadenoma de hipófisis, y el resto a hiperplasia de las células mamosomatotropas3. El exceso de GH es particularmente deletéreo en pacientes con SMA debido a que puede acelerar la displasia fibrosa, especialmente en los huesos craneofaciales, pudiendo provocar pérdida de la visión y la audición3. El tratamiento de primera línea ante el exceso de GH y la hiperprolactinemia es médico, a través del uso de análogos de somatostatina y agonistas del receptor D2 de dopamina, ya que en caso de presentarse adenomas hipofisarios el tratamiento quirúrgico (transesfenoidal) no es efectivo debido al engrosamiento masivo de la base del cráneo como consecuencia de la displasia fibrosa craneofacial3; sin embargo, cabe mencionar que la respuesta al tratamiento médico con cabergolina y octreotide LAR en este grupo de pacientes suele ser consistente pero inadecuada, no alcanzando en la mayoría de los casos los criterios de control hormonal4. De acuerdo al consenso de criterios de curación de acromegalia publicado en el año 2010, se considera que existe control de la enfermedad cuando los niveles de GH al azar son menores de 1μg/L, con niveles normales de IGF-1 para sexo y edad. En caso de discrepancias entre estas pruebas se recomienda determinar GH 2 horas sobrecarga con 75 g de glucosa cuyo valor debe ser inferior a 0,4μg/L5. Destacamos que este paciente al ser reevaluado 3 meses después de instaurada la terapéutica con octreotide LAR presentó un valor de GH basal de 2,4μg/L, con una GH a las 2 horas postcarga de 2,5μg/L, lo cual a pesar de tener un IGF-1 normal, manifestó una respuesta inadecuada al análogo de somatostatina y motivó el incremento de la dosis a 30mg mensual.
La endocrinopatía más frecuente en el SMA es la pubertad precoz, pero se han descrito de manera excepcional algunos casos de hipogonadismo central en ambos sexos6,7. En el paciente descrito, la causa debe ser adquirida, ya que presenta buena virilización y un volumen testicular adecuado. Destaca en la anamnesis funcional la hiposmia, la cual frecuentemente se asocia a hipogonadismo en el síndrome de Kallman, pero en este caso, posiblemente se deba a la esclerosis craneal severa que oblitera el cartílago cribiforme del hueso etmoides lesionando por consiguiente las fibras olfatorias. Si bien es cierto que este paciente presentaba hiperprolactinemia, sus concentraciones no eran tan elevados como para explicar el hipogonadismo, por lo cual es probable que la causa pudiera estar asociada a la lesión de las células gonadotropas debido a la displasia fibrosa de la silla turca.
En cuanto a la displasia fibrosa, esta ocurre por mutación del gen GNAS, y cursa con proliferación y diferenciación anormal de los osteoblastos en conjunto con un aumento de la actividad osteoclástica. Además, esta mutación se asocia con un aumento en la expresión del factor de crecimiento fibroblástico 23, el cual incrementa la excreción renal de fosfato, provocando hipofosfatemia y agravando el defecto de mineralización ósea2,8. Destacaremos que en este paciente se evidenció una leve hipofosfatemia al momento de su ingreso. Basado en el conocimiento fisiopatológico de la enfermedad, ha tomado vigencia en los últimos años el uso de bifosfonatos en pacientes con displasia fibrosa, siendo el pamidronato y el ácido zoledrónico los agentes más utilizados9,10. El pronóstico de la enfermedad poliostótica suele ser bueno; sin embargo, en pacientes con SMA que cursan con exceso de GH este tiende a empeorar2. El seguimiento se hace en base a marcadores de recambio óseo como la fosfatasa alcalina, los cuales están elevados en presencia de actividad de la enfermedad, y tienden a disminuir en respuesta al tratamiento antirresortivo2.
