


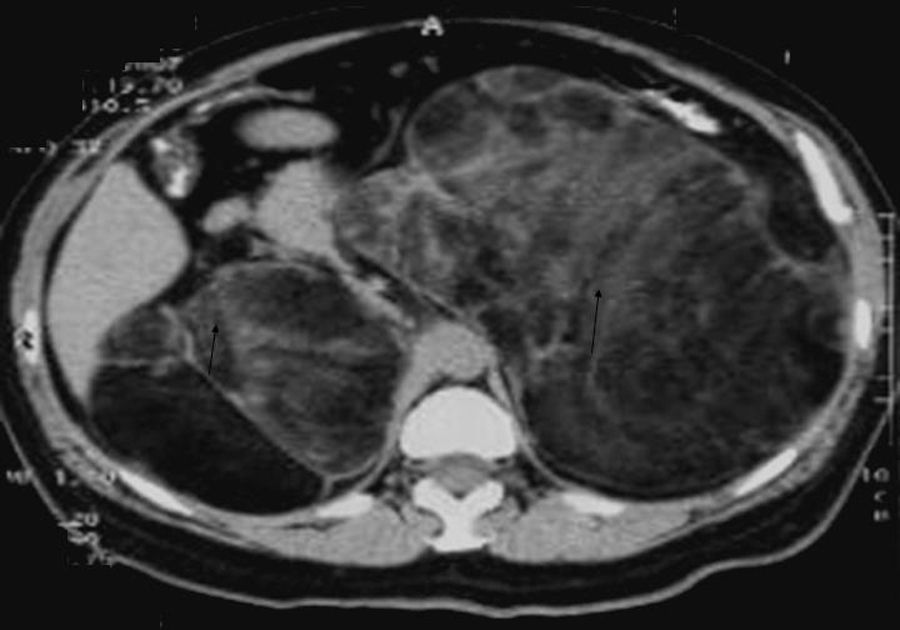
Se presenta el caso de una mujer de 64 años de edad que acude a la visita médica por un cuadro caracterizado por dolor y distención abdominal de un año de evolución. En la exploración física se encontró con una TA de 130/90mmHg, peso de 63kg, talla 1,43m, índice de masa corporal de 30,88kg/m2 y con hirsutismo a nivel abdominal sin datos de hiperpigmentación. Se solicitó una ultrasonografía abdominal en donde se evidenciaron tumoraciones en la zona retroperitoneal. Una tomografía fue solicitada para completar la valoración, la cual reportó tumores gigantes dependientes de la glándula adrenal (fig. 1). Fue sometida a una extracción de glándulas adrenales en 2 diferentes tiempos quirúrgicos mediante cirugía abierta. El resultado de la histopatología fue de mielolipoma adrenal bilateral. El izquierdo midió 21×18×7cm y el derecho midió 18×11×10cm. Inició un tratamiento de sustitución con cortisona oral y fludrocortisona después de la segunda adrenalectomía. Como antecedentes médicos importantes, al nacimiento presentó ambigüedad genital, por lo cual fue referida al hospital pediátrico en donde se le realizó el diagnóstico de hiperplasia suprarrenal congénita. En su infancia fue sometida en 2 ocasiones a cirugía de reconstrucción genital. El manejo farmacológico consistió en la administración de glucocorticoides por vía oral. El tratamiento continuó hasta los 24 años. La paciente suspendió todo medicamento y dejó de acudir a revisión médica. Permaneció 20 años asintomática sin ningún medicamento hasta la presentación del cuadro actual.

El primer punto a discutir es el diagnóstico de hiperplasia adrenal congénita que tiene la paciente. Desafortunadamente, la valoración por el Servicio de Endocrinología se solicitó después de los eventos quirúrgicos, por lo que no fue posible realizar los exámenes de laboratorio hormonales para complementar el diagnóstico. Por las características clínicas y la historia de la paciente se podrían considerar 2 tipos de bloqueo. El primero es la deficiencia de 11-hidroxilasa. Este bloqueo se caracteriza por presentar virilización en las mujeres e incremento de andrógenos. Este tipo de bloqueo generalmente no se asocia a cuadros agudos de insuficiencia adrenal por el incremento de la 11-desoxicorticoesterona, lo que a su vez conlleva en la hipertensión arterial en aproximadamente el 60% de las pacientes. En nuestro caso, la paciente jamás presentó algún evento de insuficiencia adrenal aguda. No obstante, factores en contra para pensar en este tipo de bloqueo es que su incidencia es muy baja, menor que 1 en 100.000, además de la ausencia de hipertensión arterial1. El segundo tipo de bloqueo a considerar es una deficiencia de 21-hidroxilasa virilizante, no perdedora de sal. En estos casos hay virilización en las mujeres. Pueden no presentar datos de deficiencia de mineralocorticoides porque hay producción suficiente de aldosterona. Por el momento la única manera posible de confirmar el diagnóstico es la búsqueda intencionada de mutaciones en los genes de los citocromos P 450 11 y 21. En individuos homocigotos para la forma virilizante simple de la deficiencia de 21-hidroxilasa se ha encontrado una prevalencia aproximada del 80% de incidentalomas adrenales, lo cual se cree que está en relación con la estimulación crónica por corticotropina (ACTH)2. En el caso de los mielolipomas, estos son tumores relativamente raros que están compuestos por tejido adiposo maduro y tejido hematopoyético. Se cree que provienen de células indiferenciadas del estroma. Estos tumores representan aproximadamente el 1% de todas las masas adrenales. La mayoría de estos tumores son asintomáticos y se encuentran como incidentalomas3,4,5. Se han publicado hasta la fecha aproximadamente 18 casos de pacientes con bloqueo de 21-hidroxilasa asociados a mielolipomas gigantes. En la mayoría de estos casos el diagnóstico de mielolipomas se hace en la edad adulta6,7. La fisiopatología de esta asociación no ha sido del todo explicada. Una de las posibles hipótesis que se podría plantear en nuestro caso es la ausencia por tiempo prolongado de un tratamiento con esteroides, lo que podría incrementar los valores de ACTH. Estudios en modelos animales han demostrado un aumento de la hematopoyesis en las glándulas adrenales con la administración de ACTH, lo que podría explicar esta asociación. No obstante, en casos previos no fue posible encontrar la expresión del receptor de ACTH ni de andrógenos en el tumor8. Por tanto, es difícil establecer la fisiopatología exacta de este fenómeno. La falta de tratamiento con esteroides por varios años podría ser el principal factor desencadenante de este fenómeno en nuestra paciente, así como la mayor prevalencia de tumores adrenales en pacientes con bloqueos hormonales adrenales. En conclusión, la asociación de bloqueos adrenales se asocia al incremento de tumores en la glándula. En el caso de los mielolipomas adrenales no son la excepción ya que no hay un mecanismo por el cual se pueda explicar la fisiopatología de esta asociación.
