


La acromegalia es una enfermedad caracterizada por la hipersecreción crónica de hormona de crecimiento (GH) debido a un adenoma hipofisario en más del 95% de los casos. Las alteraciones cardiovasculares, presentes hasta en el 60% de los pacientes, constituyen la principal causa de morbi-mortalidad. La apoplejía hipofisaria (AH) es el síndrome clínico que se da tras una hemorragia o infarto espontáneo de la glándula pituitaria, en general en el contexto de un adenoma preexistente. Presentamos un caso de acromegalia, diagnosticada a raíz de un episodio de insuficiencia cardíaca, que tras sufrir un episodio de AH presentó resolución de la miocardiopatía acromegálica (MA) y curación de la acromegalia.
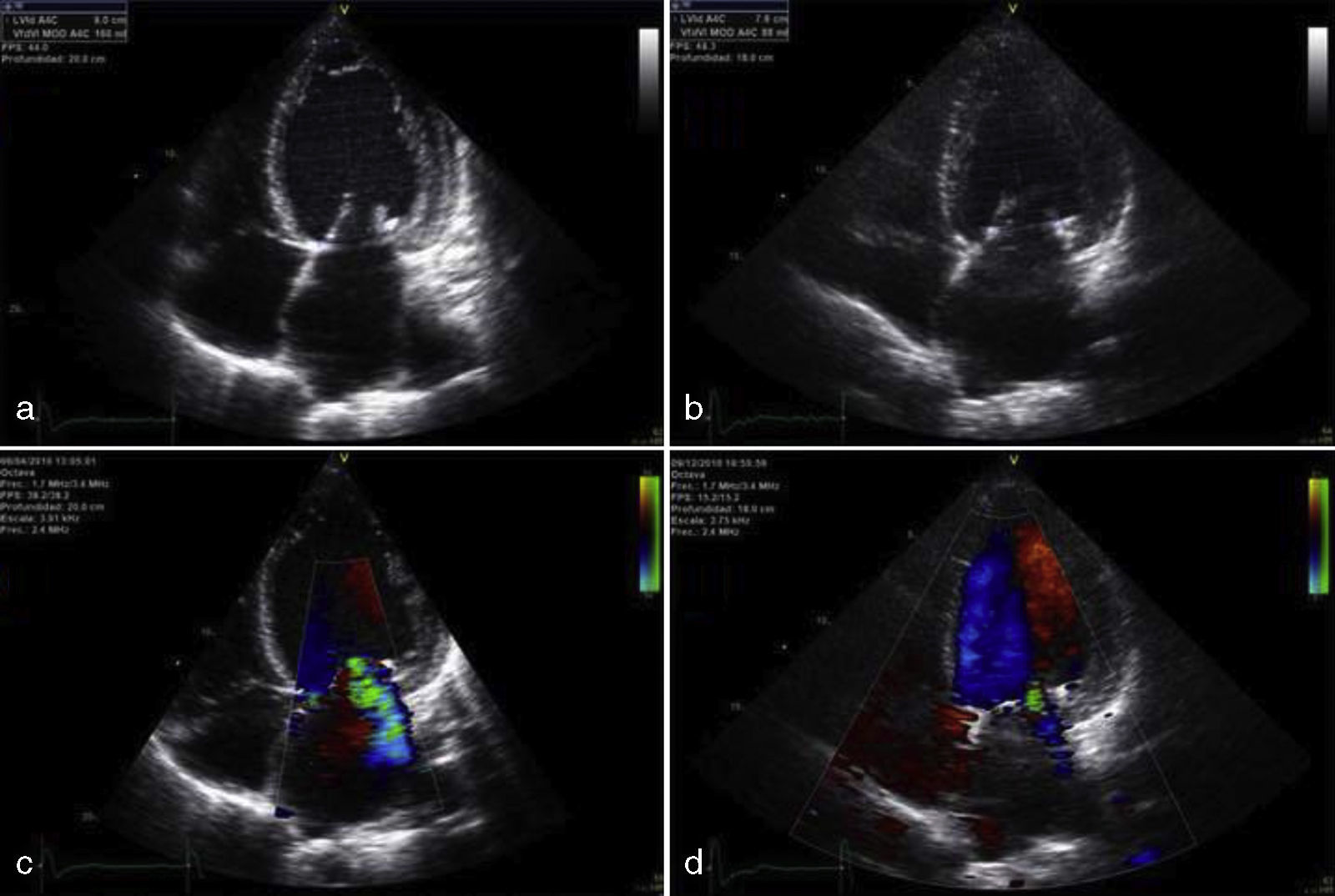
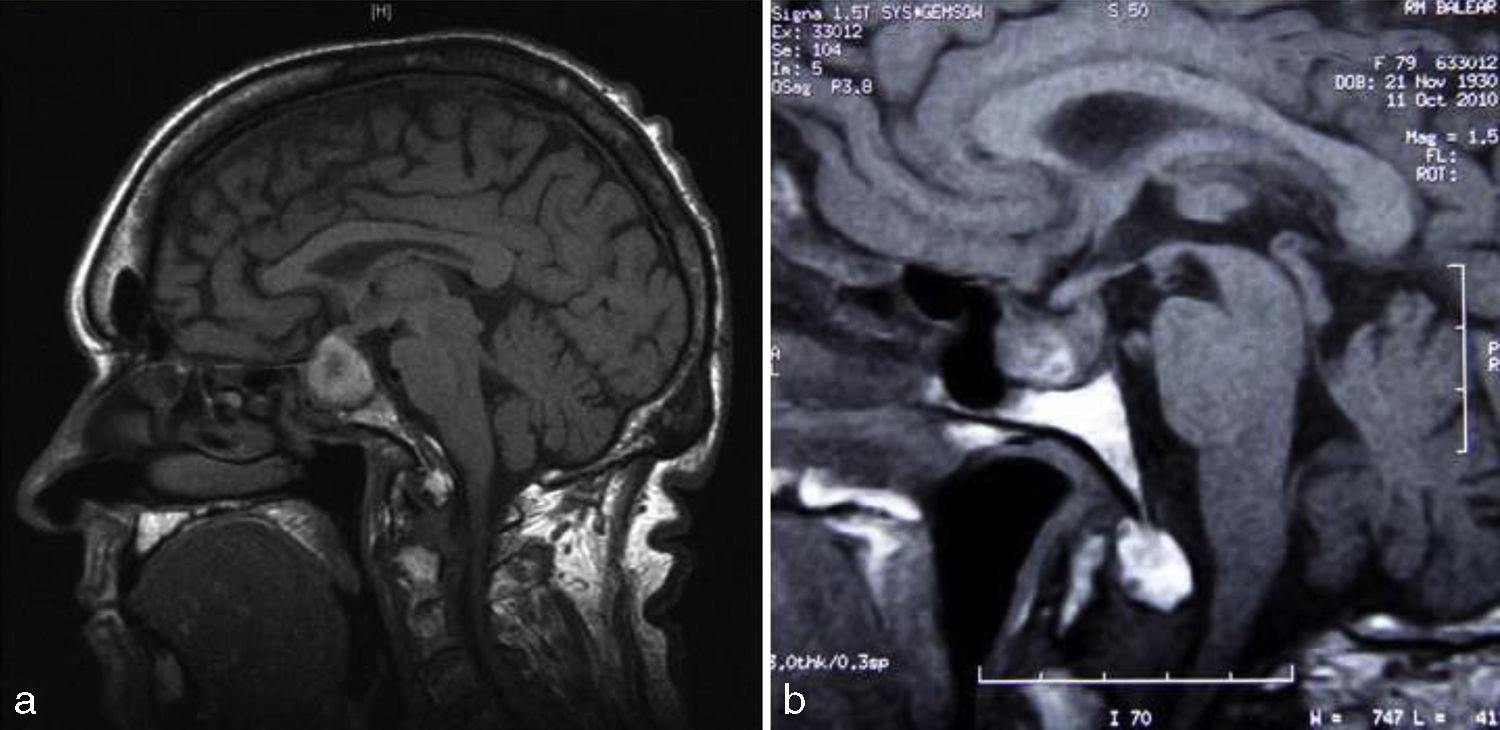
Mujer de 79 años con antecedentes de hipertensión arterial (HTA) y diabetes mellitus tipo 2 (DM2) diagnosticadas hace 15 años, con buen control y en tratamiento con amlodipino, lisinopril, hidroclorotiazida, metformina y glibenclamida. Acudió a urgencias por dolor torácico de características anginosas con bloqueo completo de rama izquierda en el ECG y sin elevación de biomarcadores de necrosis miocárdica. Ingresó con el diagnóstico de síndrome coronario agudo, iniciándose tratamiento con doble antiagregación (aspirina y clopidrogrel) y heparina de bajo peso molecular. La paciente evolucionó con disnea rápidamente progresiva, con tensión arterial de 220/120mmHg, objetivándose semiología clínica y radiológica compatible con edema agudo de pulmón. A la exploración física, el peso era de 65kg, talla de 158cm (índice de masa corporal 26kg/m2), presentaba un soplo sistólico mitral 3/6 y destacaban rasgos faciales toscos, con crecimiento de partes acras, macroglosia, prognatismo, diastema, acrocordones y bocio, todo ello sugestivo de acromegalia. La paciente refería crecimiento de las manos y aumento de 2 números del calzado desde hacía 15 años. En la analítica se comprobó glucosa: 107mg/dl, hemoglobina glucosilada: 7,9%; colesterol: 177mg/dl; triglicéridos: 64mg/dl. El ECG presentaba bloqueo completo de rama izquierda con alteraciones secundarias de la repolarización. En la radiografía de tórax se observó una cardiomegalia e infiltrados alveolo-intersticial con patrón hiliofugal bilateral. El ecocardiograma transtorácico mostraba aurícula izquierda dilatada de 56,4ml/m2, ventrículo izquierdo dilatado de 70mm, ligeramente hipertrófico, con una disfunción sistólica ventricular avanzada (fracción de eyección calculada por Simpson biplano del 20%) por hipocinesia moderada del septo y grave del resto de los segmentos, insuficiencia mitraliii/iv e insuficiencia tricúspidea i/iv (fig. 1a). Tras evolución favorable y con la sospecha de MA, se solicitó estudio hormonal y resonancia magnética nuclear (RM) hipofisaria. La paciente desarrolló un episodio de cefalea brusca, acompañada de náuseas, vómitos, disminución de la agudeza visual y tensión arterial de 50/30mmHg con afectación del estado general. La analítica mostró hiponatremia de 127 mEql/L. Ante la sospecha de AH e insuficiencia suprarrenal secundaria, se instauró tratamiento con glucocorticoides endovenosos. La RM hipofisaria reveló una lesión intraselar de 2,3×2,3cm con imágenes sugestivas de hemorragia que contactaba con el quiasma óptico, compatible con apoplejía de un macroadenoma (fig. 2a). La campimetría practicada no fue valorable. El estudio hormonal mostró hipogonadismo hipogonadotropo con homona folículoestimulante: 0,4 mUI/ml (23-116), hormona luteinizante indetectable (5,9-54) y estradiol: 12,6pg/ml (0-37); insuficiencia suprarrenal secundaria con cortisol: 1,52μg/dl (4,3-25) y hormona corticotropa: 6,9pg/ml (1-46); prolactina: 11,2ng/ml (1,8-20,3); tirotropina: 0,07μUI/ml (0,35-5,5), tiroxina no unida a proteína: 1,25ng/dl (0,89-1,76) y triyodotironina no unida a proteína: 1,61pg/ml (2,3-4,2), sugestivo de síndrome de eutiroideo enfermo; GH: 1,07ng/ml (0,05-7,4), factor de crecimiento insulínico tipo I (IGF-I): 120ng/ml (55-166). No se disponía de valores previos de GH e IGF-I. Fue dada de alta con metformina, enalapril, carvedilol, furosemida, espironolactona e hidrocortisona. Tras 8 meses de seguimiento, el ecocardiograma transtorácico mostró una disminución del diámetro telediastólico ventricular izquierdo con incremento de la fracción de eyección (50% Simpson biplano) y disminución de la insuficiencia mitral (fig. 1b). En la RM hipofisaria realizada a los 6 meses, se observó una reducción del tamaño tumoral (1,1×1,7cm) con menos contenido hemorrágico (fig. 2b). Persistía hipogonadismo hipogonadotro e insuficiencia suprarrenal secundaria. Presentaba hipertiroidismo subclínico (tirotropina: 0,03,: tiroxina no unida a proteína 1,54) debido al bocio, prolactina normal; GH: 0,54ng/ml e IGF-I: 40,6ng/ml, hemoglobina glucosilada de 5,6% y campimetría normal.
 que se normaliza a los 8 meses de la apoplejía hipofisaria (figura b) mejorando la fracción de eyección. Nótese la disminución en la gravedad de la insuficiencia mitral antes (figura c) y después de la apoplejía hipofisaria (figura d).")
Imagen ecocardiográfica desde el plano apical de 4 cámaras que muestra ventrículo izquierdo con volúmen telediastólico aumentado (figura a) que se normaliza a los 8 meses de la apoplejía hipofisaria (figura b) mejorando la fracción de eyección. Nótese la disminución en la gravedad de la insuficiencia mitral antes (figura c) y después de la apoplejía hipofisaria (figura d).
. Disminución del tamaño tumoral a los 6 meses de seguimiento (figura b).")
En la MA existen alteraciones morfológicas y funcionales debidas al exceso crónico de GH e IGF-I. En la etapa temprana de la enfermedad, el efecto trófico e inotrópico de la GH provoca una hipertrofia miocárdica con afectación biventricular característica de la MA y un síndrome hiperdinámico. La MA evoluciona con hipertrofia ventricular más grave y se desarrolla fibrosis intersticial, que conduce a una disfunción diastólica y una intolerancia al ejercicio1. En la última etapa, existe un aumento de la apoptosis y fibrosis miocárdica con dilatación ventricular, disfunción sistólica y diastólica pudiendo aparecer alteración valvular funcional, siendo frecuente la insuficiencia mitral2,3. La edad del paciente y la larga exposición al exceso de GH e IGF-I son los principales determinantes del desarrollo de la MA, presente hasta en el 90% de los pacientes4. La insuficiencia cardíaca se observa en el 3-10% de los pacientes siendo más significativa en pacientes con acromegalia de larga evolución, HTA y disfunciones valvulares5. Existen una serie de factores asociados a la progresión de la MA como HTA, DM2, hipertrigliceridemia, tirotoxicosis, enfermedad arterial coronaria, presencia de arritmias y enfermedades respiratorias. El diagnóstico diferencial de la MA se establece fundamentalmente con la miocardiopatía dilatada de origen isquémico, hipertensivo o idiopático. En el caso descrito, la ausencia de elevación de marcadores de necrosis miocárdica así como de alteraciones de la motilidad segmentaria ventricular izquierda hacían que el origen isquémico de la miocardiopatía fuera poco probable. Se consideró la posibilidad de una cardiopatía hipertensiva en fase dilatada pero la paciente presentaba una historia de HTA de larga evolución con buen control bajo adecuado tratamiento farmacológico. La MA suele presentar una importante regresión y en algunos casos resolución tras curación de la acromegalia, como ocurre en el caso, lo que apoya su diagnóstico.
El tratamiento de la acromegalia incluye cirugía transesfenoidal, tratamiento farmacológico (análogos de somatostatina, antagonistas del receptor de GH, agonistas dopaminérgicos) y radioterapia6. Es excepcional la curación tras un episodio de AH. La AH ocurre en el 0,6-10% de los adenomas hipofisarios y la presentación clínica varía desde formas asintomáticas hasta emergencias vitales7. Las manifestaciones clínicas más frecuentes son cefalea, vómitos, afectación de campo visual y de pares craneales. La AH puede provocar cualquier déficit hormonal hipofisario transitorio o permanente. Los mecanismos implicados no están claramente establecidos. Las anomalías vasculares del tumor, la isquemia debido al rápido crecimiento del adenoma y la compresión de la arteria hipofisaria superior contra el diafragma selar son mecanismos que contribuyen a la apoplejía. La edad avanzada puede incrementar la fibrosis pituitaria y fragilidad de los vasos. La DM2, puede contribuir al desarrollo de un infarto debido a los cambios degenerativos en la microcirculación hipofisaria que provoca7,8. Entre los posibles desencadenantes se encuentran los test de estimulación con hormonas hipotalámicas, tratamiento anticoagulante y antiagregante, cambios en la tensión arterial, meningitis, contrastes radiológicos, procedimientos quirúrgicos, trauma craneal y algunos fármacos (atenolol y butformina). El tratamiento de la AH se basa en la descompresión quirúrgica y terapia sustitutiva hormonal. El abordaje inicial comprende medidas de soporte vital, reposición hidroelectrolítica, analítica para evaluar los déficits hormonales y dosis elevadas de corticoides, pues la insuficiencia suprarrenal secundaria es la causa de mayor morbi-mortalidad. La descompresión quirúrgica se recomienda de forma temprana cuando existe disminución de la agudeza visual, déficit severo y persistente del campo visual y deterioro del nivel de consciencia. La reposición hormonal sustitutiva dependerá de los déficits asociados, quedando un hipopituitarismo en el 50-87% de los casos9,10. Fraser et al. presentan 22 casos de remisión de acromegalia tras apoplejía hipofisaria. Ocho casos ocurrieron en pacientes sin previo diagnóstico formal de acromegalia, la cefalea grave retroorbitaria fue el síntoma más frecuente. Hubo normalización de GH en 19 pacientes. La apoplejía causó déficit hormonal en 15 pacientes y en 2 pacientes ocasionó una mejoría o normalización de un déficit previo. Hubo 7 casos de mejoría o curación de DM2 y un caso de mejoría de la HTA7.
Nuestra paciente presenta una MA en fase avanzada, debido a una acromegalia de larga evolución no diagnosticada, edad avanzada, y 2 factores que pueden haber influido en la progresión de la miocardiopatía como son la HTA y la DM2. El tratamiento con doble antiagregación, anticoagulación y cambios en la tensión arterial pudieron desencadenar la apoplejía. Se produjo una insuficiencia suprarrenal secundaria y un déficit de gonadotrofinas. La AH ocasionó curación de la acromegalia lo que junto con el tratamiento médico específico (betabloqueantes, inhibidores de la enzima conversora de angiotensina, antialdosterónicos), provocó una regresión y práctica resolución de la MA tras 8 meses de seguimiento. Se produjo una mejoría de la DM2, en tratamiento actual con metformina y hemoglobina glucosilada de 5,6%.
El diagnóstico y tratamiento precoz de la acromegalia junto con el control de otros factores de riesgo cardiovasculares son importantes para reducir la morbi-mortalidad cardiovascular asociada y mejorar la calidad de vida.
El abordaje multidisciplinar del paciente acromegálico requiere el conocimiento de los síntomas asociados a una AH, así como los factores desencadenantes y el tratamiento por parte de los clínicos implicados.
El seguimiento de los pacientes que han presentado una AH es importante para detectar nuevos déficits hormonales o recurrencia de la enfermedad, que puede ocurrir incluso varios años después del episodio.
