


El síndrome de Boucher-Neuhäuser (SBN) es una condición clínica caracterizada por la tríada de hipogonadismo hipogonadotrópico, ataxia cerebelosa y degeneración retinocoroidal1,2. En etapas tempranas, esta patología resulta de difícil diagnóstico ya que los componentes de la tríada se desarrollan en diferentes momentos de la vida de los pacientes con una amplia variabilidad entre los pocos sujetos descritos hasta la fecha3.
Presentamos el caso de un paciente varón de 26 años de edad, natural y procedente del Estado Mérida (Venezuela) sin antecedentes familiares de consanguinidad ni enfermedades importantes en sus padres y hermanos, quien fue referido a la Unidad de Endocrinología por ausencia de caracteres sexuales secundarios, refiriendo además disminución de agudeza visual bilateral a partir de los 16 años y marcha inestable desde los 20 años.
El examen físico reveló peso corporal de 57 kg, talla 160,0cm, brazada 165,7cm, índice de masa corporal de 22,2 kg/m2, hábito eunucoide, voz de tonalidad aguda, ginecomastia bilateral, nistagmus horizontal bilateral con fase rápida a la derecha, agudeza visual corregida de ojo derecho que contaba dedos a 50cm, y ojo izquierdo 20/80, presión intraocular de 12mmHg en ambos ojos, vello púbico Tanner II, pene de 4cm de longitud, testículo derecho de 2mL e izquierdo de 3mL (orquidómetro de Prader) ambos en bolsas escrotales hipoplásicas. El examen neurológico reveló marcha atáxica, ataxia troncal a predominio derecho, postura distónica, disdiadococinesia, dismetría, hipotonía e hiporeflexia global. No se encontraron alteraciones en la sensibilidad superficial ni profunda, tampoco anosmia ni alteraciones cardiovasculares.
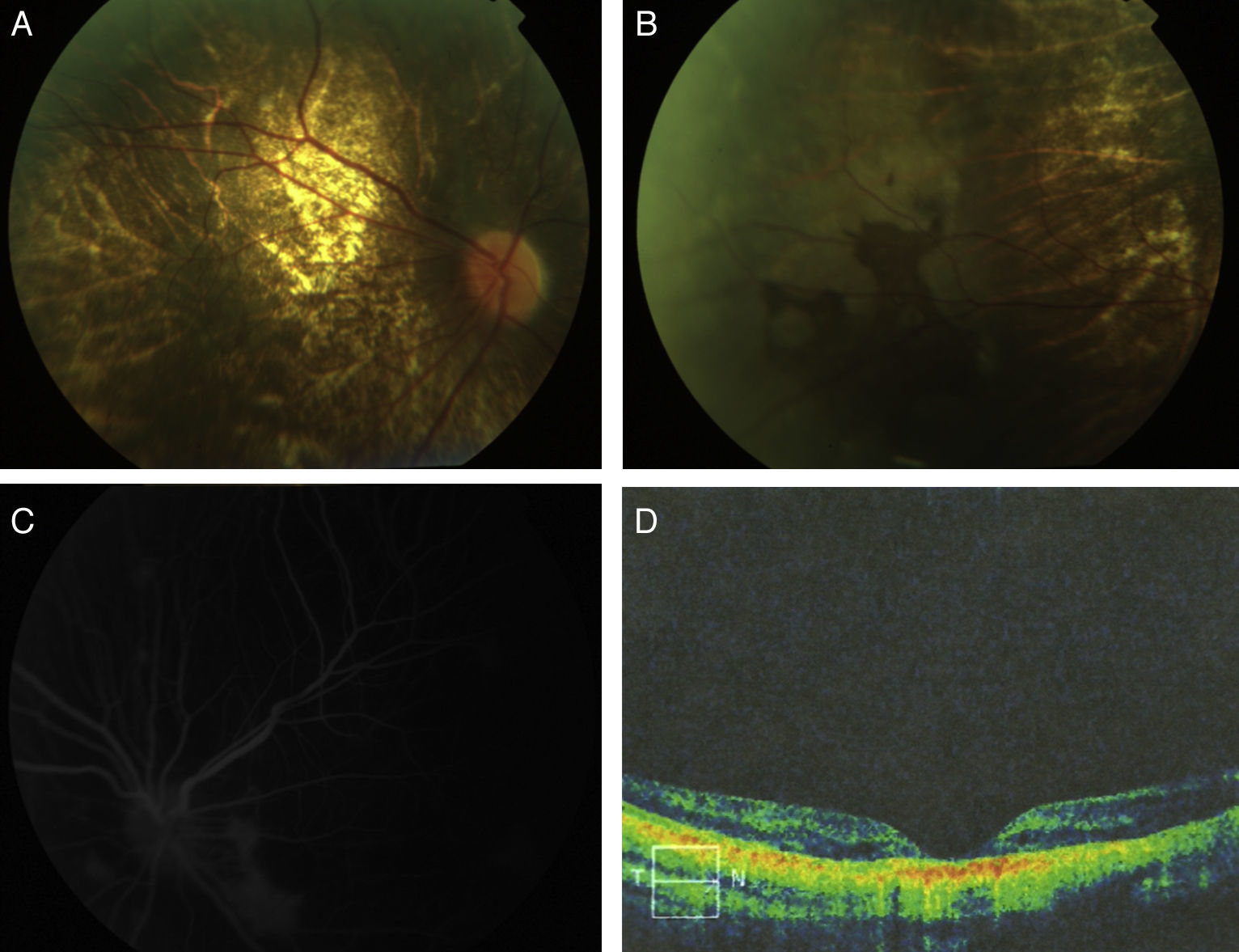
El examen del fondo ocular evidenció en ambos ojos un disco óptico pálido, vasos retinianos con leve tortuosidad y distrofia retinocoroidal caracterizada por atrofia difusa del epitelio pigmentario de la retina, con depósito de pigmento hacia la periferia y visualización de vasos coroideos. La angiografía con flouresceína demostró en ambos ojos un árbol vascular conservado con atrofia difusa del epitelio pigmentario de la retina y zonas hiperfluorescentes con acúmulo de colorante. Se realizó tomografía de coherencia óptica, apreciándose adelgazamiento retiniano con alteraciones de las capas externas y un epitelio pigmentario engrosado e irregular de predominio foveal que afectaba la coriocapilaris (fig. 1).
 Fotografía del fondo ocular donde se evidencia un disco óptico pálido, vasos retinianos con leve tortuosidad y distrofia retinocoroidal caracterizada por atrofia difusa del epitelio pigmentario de la retina y visualización de vasos coroideos. B) Fotografía del fondo ocular en la que se demuestra el depósito de pigmento hacia la periferia. C) Angiografía con fluoresceína en la que se observa un árbol vascular conservado y zonas hiperfluorescentes de acúmulo de colorante. D) Tomografía de coherencia óptica en la que se evidencia adelgazamiento retiniano con alteración de las capas externas de la retina.")
A) Fotografía del fondo ocular donde se evidencia un disco óptico pálido, vasos retinianos con leve tortuosidad y distrofia retinocoroidal caracterizada por atrofia difusa del epitelio pigmentario de la retina y visualización de vasos coroideos. B) Fotografía del fondo ocular en la que se demuestra el depósito de pigmento hacia la periferia. C) Angiografía con fluoresceína en la que se observa un árbol vascular conservado y zonas hiperfluorescentes de acúmulo de colorante. D) Tomografía de coherencia óptica en la que se evidencia adelgazamiento retiniano con alteración de las capas externas de la retina.
En los análisis de laboratorio no se encontraron alteraciones hematológicas; sin embargo, se observó hipersegmentación de neutrófilos en frotis de sangre periférica. La bioquímica sanguínea demostró valores normales de glucemia, función renal, calcio y fósforo, hormona folículo estimulante (FSH): 0,5 mUI/mL (valores normales [VN]: 0,7-11), hormona luteinizante (LH): 0,3 mUI/mL (VN: 0,8-7,6), testosterona total: 0,9 ng/mL (VN: 2,45-18), prolactina: 8,4 ng/mL (VN hombres: 0-15), tirotropina: 1,2 mUI/mL (VN: 0,3-4,2), tiroxina libre: 0,9 ng/dL (VN: 0,8-2). Las concentraciones plasmáticas de LH y FSH no aumentaron a los 30, 60 y 90 minutos después de la administración intravenosa de 100 ug de hormona liberadora de gonadotrofinas (GnRH).
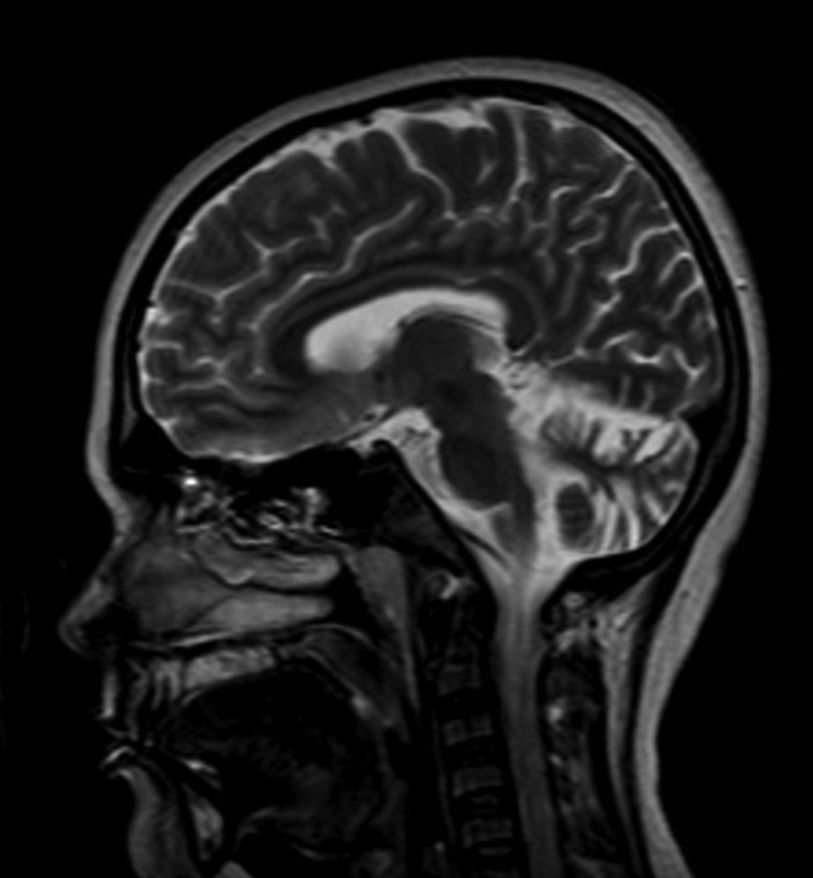
En 50 metafases analizadas el cariotipo fue 46, XY; la radiografía de mano y muñeca izquierda reveló una edad ósea de 16 años de acuerdo al atlas de Greulich y Pyle. En la resonancia magnética cerebral se evidenció un aumento en la amplitud y profundidad de ambos hemisferios cerebelosos compatible con atrofia cerebelosa (fig. 2). La densitometría ósea mostró una densidad mineral ósea de L1-L4 de 0,749g/cm2, que relacionado con la población de la misma edad cronológica (26 años) corresponde a un Z-score de -3,1 desviación estándar (DS), y respecto a la población de la misma edad ósea (16 años) el Z-score era de -2,5 DS. A nivel de cuello femoral total la masa ósea fue de 0,687g/cm2, lo cual corresponde a un Z-score de -2,1 DS y -1,9 DS relacionado con la población de 26 y 16 años, respectivamente. Asimismo, la densidad mineral ósea en cuerpo total fue de 1,047g/cm2 que corresponde a un Z-score de -1,7 DS (26 años) y -1,2 DS (16 años).

Con el diagnóstico de hipogonadismo hipogonadotrópico, se indicó enantato de testosterona de depósito a dosis de 250mg intramuscular cada 21 días, y para la osteoporosis teriparatide (hormona paratiroidea humana [1-34]) 20μg subcutáneo al día, y suplementos de calcio y vitamina D.
Las características principales del paciente que presentamos son: distrofia retinocoroidal, hipogonadismo hipogonadotrópico y ataxia cerebelosa. Dicha tríada ha sido descrita previamente y es compatible con el SBN1–3. La prevalencia de esta patología se desconoce, pero hasta el año 1997 solo se habían descrito 17 casos en todo el mundo, siendo afectados en proporciones iguales, varones y mujeres3.
El compromiso ocular puede ser la primera manifestación de la enfermedad, lo cual ocurre entre la primera y la sexta década de la vida. De igual forma, la expresión y progresión de la degeneración retinocoroidal varía ampliamente en los casos publicados, pero se ha descrito que la principal manifestación ocular viene dada por la atrofia difusa del epitelio pigmentario de la retina con depósito de pigmento en el polo posterior y la periferia4, tal como se encontró en nuestro paciente.
Los hallazgos neurológicos más frecuentemente encontrados en el SBN son marcha inestable, nistagmus, dismetría y disdiadococinesia, los cuales configuran un síndrome cerebeloso atribuido a la atrofia significativa de dicha estructura3. Al igual que en este paciente, se han descrito algunos casos que cursan con hiporeflexia global y un grado variable de hipotonía muscular sin compromiso de la sensibilidad3. La clínica neurológica es con frecuencia no progresiva y se presenta de forma tardía, después de los 15 años de edad1–3.
En cuanto a la esfera endocrinológica, la ausencia de caracteres sexuales secundarios constituye el principal motivo de consulta de estos pacientes, sugiriendo un estado hipogonadal, el cual en el SBN corresponde a un hipogonadismo hipogonadotrópico. En algunos casos como consecuencia de la deficiencia de GnRH5, mientras que en otros (como en el presente caso), la ausencia de respuesta al estímulo con GnRH ha sugerido disfunción de las células gonadotropas3. En todo caso, el déficit androgénico, además de comprometer la fertilidad de los afectados, también altera el metabolismo óseo, disminuyendo la masa ósea, creando un riesgo importante de fracturas vertebrales y no vertebrales. Recientemente, el Grupo de Trabajo de Metabolismo Mineral de la Sociedad Española de Endocrinología y Nutrición (SEEN) ha propuesto una serie de recomendaciones prácticas para la evaluación y tratamiento de la osteoporosis asociada a diferentes enfermedades endocrinas, y en el caso de la osteoporosis secundaria a hipogonadismo masculino recomiendan restaurar los niveles de testosterona, mantener un nivel adecuado de calcio y vitamina D y realizar actividad física regular6. Además, en pacientes que cursan con masa ósea muy baja (<-3DS) recomiendan como opción terapéutica el uso de teriparatide6. Al respecto, Orwoll et al.7 han demostrado que la administración diaria subcutánea de 20 o 40μg de teriparatide incrementa significativamente la densidad mineral ósea de columna y cuello femoral tanto en hombres hipogonadales como eugonadales con osteoporosis. De igual forma, el tratamiento sustitutivo con testosterona en adolescentes y adultos jóvenes con hipogonadismo hipogonadotrópico idiopático se ha asociado con un incremento en la masa ósea tanto cortical como trabecular8.
En los últimos años se ha descrito el fenómeno de hipersegmentación de neutrófilos en sujetos con SBN, el cual se define como la presencia de al menos 5% de neutrófilos con 5 o más lóbulos9. Este hallazgo hematológico observado en este paciente refuerza la hipótesis de que la hipersegmentación de neutrófilos es una característica común en este síndrome, y que la misma no causa aparente disfunción inmunológica. Además, el paciente no presenta anemia megaloblástica, uremia, infecciones, fiebre o enfermedad renal crónica, las cuales son causas de hipersegmentación.
A pesar de que los tejidos implicados en la tríada son de origen neuroectodérmico, la relación exacta entre los 3 desórdenes no se conoce. Limber et al.10 en el año 1989 reevaluaron uno de los pacientes descritos inicialmente por Neuhäuser y Opitz, y sugirieron que el defecto subyace en un gen único con efectos pleiotrópicos, pero la naturaleza del gen y su papel en la fisiopatología de la enfermedad se desconoce. La ausencia de manifestaciones clínicas similares en los hermanos del paciente sugiere que el patrón de herencia es autosómico recesivo como ha sido descrito previamente2. Actualmente no existe una prueba genética para identificar sujetos con SBN. Por lo tanto, su diagnóstico es clínico y debe realizarse con base en la tríada clásica.
Los autores desean agradecer a los doctores Rafael Muci-Mendoza, Gustavo Paredes, Daniela Hernández y Luís Betancourt por su asesoría en la evaluación integral del paciente, así como al Laboratorio Bioclínico Glorias Patrias por su colaboración en la realización de los estudios hormonales.
