


La adrenoleucodistrofia ligada al cromosoma X (ALD-X) es la enfermedad peroxisomal más frecuente, con una incidencia estimada de 1/40.000 nacimientos y afectación casi exclusiva de varones1. Dada la ausencia de un tratamiento específico eficaz, durante años se ha empleado el aceite de Lorenzo como terapia común para todos los afectos1. Hoy en día, parece que su característica más prometedora es su posible capacidad preventiva, siendo ineficaz una vez manifestada la enfermedad1–4.
Paciente de 18 años con hemofilia tipo A, diagnosticado de ALD-X a los 2 años de edad en Portugal. Entre sus antecedentes familiares destacaba la existencia de hemofilia en la rama materna, así como un primo materno fallecido de ADL-X. A pesar de las recomendaciones, su madre había rechazado el consejo genético prenatal. A los 4 años de edad inició tratamiento con aceite de Lorenzo (35ml/día) que mantuvo únicamente durante 1 mes. Posteriormente, por traslado de domicilio, este fue suspendido. Desde los 4 hasta los 7 años se realizaron resonancias magnéticas (RM) cerebrales y estudios de la función adrenal periódicos en el Servicio de Pediatría que resultaron siempre normales. A los 7 años, abandonó el seguimiento. El paciente permaneció asintomático y con buen desarrollo físico, psíquico y neurológico hasta los 20 años. Llegada esa edad, acudió a urgencias por un episodio sincopal y una crisis convulsiva parcial con pruebas de imagen y electroencefalograma (EEG) normal. Dos años después, sufrió varias crisis convulsivas tónico-clónicas generalizadas que requirieron ingreso. En el interrogatorio dirigido, el paciente refirió dificultad para la lectura y comprensión de meses de evolución y alteraciones del equilibrio. El EEG confirmó la existencia de una focalidad temporal izquierda y en la RM se apreció enfermedad de la sustancia blanca subcortical bilateral con alteración de la mielinización, secundarias a ADL-X. El estudio analítico realizado confirmó la alteración del perfil lipídico con acumulación de ácidos grasos saturados de cadena muy larga (AGCML): C22:0: 2,15 (normal: 0,18-0,48); Cociente C24:0/C22:0: 1,45 (normal <1); Cociente C26:0/22:0: 0,1 (normal <0,02). Se inició tratamiento con ácido valproico (500mg/24 horas) y se solicitó interconsulta al Servicio de endocrinología y nutrición para estudio hormonal y valoración de tratamiento con aceite de Lorenzo. La exploración física mostró: peso 68kg, talla 172cm, IMC 23,3 Kg/m2, presión arterial 120/75mmHg. El resto de la exploración resultó anodina sin datos sugestivos de insuficiencia suprarrenal ni hipogonadismo. El estudio hormonal fue normal. La encuesta dietética mostró una ingesta variada sin restricción de alimentos. Dadas las manifestaciones neurológicas presentes, la ausencia de eficacia demostrada en estas situaciones y los posibles efectos adversos (trombopenia) en un paciente con sus características de base (hemofilia) no se consideró indicado el tratamiento con aceite de Lorenzo. Actualmente se está valorando el posible beneficio/riesgo de un transplante de precursores hematopoyéticos (TPH).
El principal defecto metabólico de la ALD-X consiste en una acumulación de AGCML en los tejidos, principalmente en la sustancia blanca cerebral, córtex adrenal y testículos. Esto se debe a distintas mutaciones del gen ABCD1, que codifica una proteína de la membrana de los peroxisomas (ALDP) cuya función es intervenir en la degradación por betaoxidación de estos ácidos grasos1. Su diagnóstico definitivo se realiza mediante la confirmación del incremento de AGCML (concretamente el C26:0, C24:0 y C22:0) en plasma y cultivos de fibroblastos epiteliales1. De ellos, el C26:0 es el más consistentemente elevado y por ello se considera diagnóstico de la enfermedad. Aunque pacientes con dietas cetogénicas pueden presentar también elevaciones de AGCML, estas suceden únicamente en plasma y no en fibroblastos1. Además, la determinación de estos AGCML y de ácidos poliinsaturados, permite el diagnóstico de carencias nutricionales a lo largo del seguimiento. El estudio bioquímico de familiares es crucial, ya que permite diagnosticar a varones hemicigotos presintomáticos y ofrecer consejo genético a las mujeres heterocigotas.
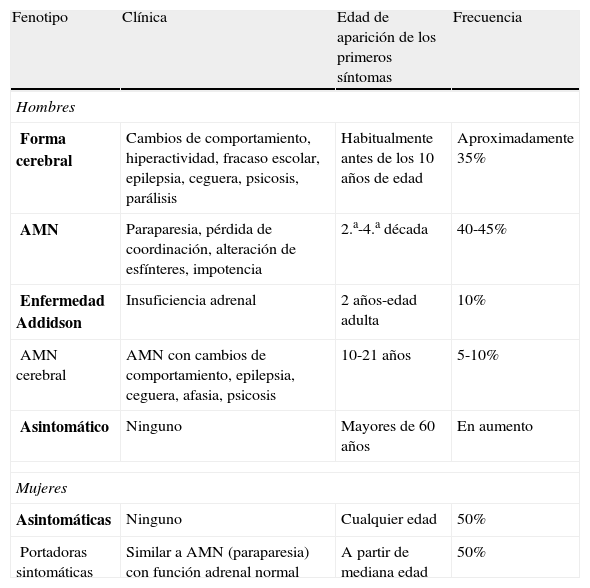
Existen varios fenotipos (tabla 1)5: la forma cerebral (infantil, del adolescente o de la edad adulta), una forma de paraparesia lentamente progresiva del adulto (AMN) e incluso individuos asintomáticos. La afectación adrenal puede presentarse a cualquier edad independientemente del tipo de afectación neurológica. De hecho, el 10% de los pacientes presentan exclusivamente síndrome de Addison1. Además, en los adultos puede existir disfunción testicular con hipogonadismo e impotencia.
Fenotipos de ALD-X
| Fenotipo | Clínica | Edad de aparición de los primeros síntomas | Frecuencia |
| Hombres | |||
| Forma cerebral | Cambios de comportamiento, hiperactividad, fracaso escolar, epilepsia, ceguera, psicosis, parálisis | Habitualmente antes de los 10 años de edad | Aproximadamente 35% |
| AMN | Paraparesia, pérdida de coordinación, alteración de esfínteres, impotencia | 2.a-4.a década | 40-45% |
| Enfermedad Addidson | Insuficiencia adrenal | 2 años-edad adulta | 10% |
| AMN cerebral | AMN con cambios de comportamiento, epilepsia, ceguera, afasia, psicosis | 10-21 años | 5-10% |
| Asintomático | Ninguno | Mayores de 60 años | En aumento |
| Mujeres | |||
| Asintomáticas | Ninguno | Cualquier edad | 50% |
| Portadoras sintomáticas | Similar a AMN (paraparesia) con función adrenal normal | A partir de mediana edad | 50% |
AMN: paraparesia lentamente progresiva del adulto.
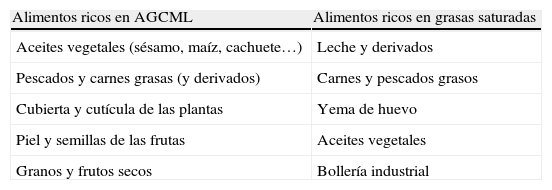
En su manejo, consideraremos 2 situaciones: 1) tratamiento preventivo (dieta y aceite de Lorenzo) y 2) tratamiento sintomático. El objetivo de la dieta es la restricción de AGCML y grasas saturadas, para evitar su acumulación (tabla 2). Para ello se proporcionan 5-10mg/día de C26:0 y el aporte de grasas se limita a un 15% del aporte calórico total. Para asegurar el cumplimiento de estas recomendaciones, es fundamental la presencia de un equipo multidisciplinar que cuente con la presencia de una dietista que impartirá una educación dietética exhaustiva al paciente y sus familiares y supervisará y asesorará en la elaboración de unos menús adecuados y más palatables. Como la disminución de la ingesta no es suficiente para descender los niveles de AGCML, se asocia el denominado aceite de Lorenzo (GTO/GTE) (mezcla de glicerol trioleato [GTO] y glicerol trierucato [GTE] en una proporción 4/1). Este disminuye o normaliza las concentraciones de AGCML. Sin embargo, hoy día sabemos que los niveles de AGCML no se correlacionan con el grado de afectación neurológica y por tanto la normalización de sus concentraciones no puede utilizarse como marcador de éxito terapéutico. Diversas publicaciones (con diseño y resultados controvertidos) apuntan a que la administración de GTO/GTE en pacientes asintomáticos podría prevenir la aparición de síntomas neurológicos2,4–8, aunque se precisan más estudios. Así, su utilización parece indicada en pacientes neurológicamente asintomáticos, preferentemente menores de 8-10 años con RM normal. En mayores de esta edad, generalmente no se recomienda su empleo porque el riesgo de desarrollar la forma cerebral es bajo. Además, dado que puede disminuir los niveles de ácidos grasos esenciales y particularmente de ácido docosahexanoico, tampoco se recomienda en menores de 1 año. En la AMN no se ha evidenciado ningún beneficio, si bien en caso de haberse iniciado y ser bien tolerado, podría continuarse. Otro tema controvertido, además de la indicación o no del aceite de Lorenzo, es también su duración. Dada la ausencia de estudios a largo plazo (con más de 10 años de seguimiento) que evalúen este aspecto, esta deberá individualizarse. En general, y dado que sus posibles efectos beneficiosos desaparecen tras su suspensión, el tratamiento puede mantenerse de forma indefinida, suspendiéndose en caso de progresión de la enfermedad o aparición de efectos secundarios graves.
Principales alimentos a evitar en la ADL-X
| Alimentos ricos en AGCML | Alimentos ricos en grasas saturadas |
| Aceites vegetales (sésamo, maíz, cachuete…) | Leche y derivados |
| Pescados y carnes grasas (y derivados) | Carnes y pescados grasos |
| Cubierta y cutícula de las plantas | Yema de huevo |
| Piel y semillas de las frutas | Aceites vegetales |
| Granos y frutos secos | Bollería industrial |
El GTO/GTE se administra en dosis de 2-3ml/kg/día, para cubrir el 20% de las calorías diarias necesarias. Habitualmente es bien tolerado, siendo sus efectos secundarios más graves la trombocitopenia9 (resuelta al suspender el tratamiento) y la linfopenia10. Por este motivo, trimestralmente se recomienda la realización de un hemograma. Además, semestralmente se precisa la valoración de enzimas hepáticas, AGCML, monoinsaturados y poliinsaturados acompañada de una detallada exploración neurológica, neuropsicológica/inteligencia, endocrinológica (monitorizando la función adrenal) y realización de potenciales evocados.
Una vez presente la afectación neurológica, el GTO/GTE no es eficaz para detener la progresión de la enfermedad ni remitir la sintomatología1–5. Se ha especulado que esto podría deberse a que, de forma similar a la fenilcetonuria, no exista respuesta a la corrección de la alteración bioquímica una vez instaurado el daño neurológico o a que tal vez el GTO/GTE disminuya el AGCML en plasma pero no en cerebro11. Tras la publicación de los últimos estudios controlados y aleatorizados, también parece descartado el posible beneficio clínico de la lovastatina12 y de tratamientos inmunosupresores o inmunomoduladores como la ciclofosfamida, interferón beta o inmunoglobulinas intravenosas13. En esta situación, el único tratamiento que podría ser efectivo en la actualidad (siempre y cuando exista un donante compatible) es el TPH realizado en fase de enfermedad cerebral precoz, aunque este no corrige la insuficiencia suprarrenal en caso de existir14. Además, recientemente15 se ha evaluado que la combinación de aceite de Lorenzo con suplementación de ácido docosahexanoico parece aumentar los niveles de ácido docosahexanoico en plasma y glóbulos rojos, pudiendo ejercer un mecanismo antiinflamatorio neuroprotector. En los próximos años, se esperan novedades terapéuticas en el manejo de esta enfermedad, dada la existencia de prometedores tratamientos actualmente en evaluación13 como son: a) el autotransplante de médula ósea (útil en ausencia de donante compatible) en combinación con terapia génica (en este caso las células de médula ósea serían corregidas genéticamente ex vivo, transfiriéndoles el gen ABCD1 con ayuda de un vector lentiviral derivado del virus de inmunodeficiencia adquirida modificado e inactivado); b) la inducción farmacológica de ABCD2 (el transportador peroxisomal más homólogo al ABCD1) mediante inhibidores de la histona deacetilasa como el ácido valproico (con efectos antioxidantes añadidos) o el 4-fenilbutirato (en estos pacientes la inducción del ABCD2 podría compensar la carencia funcional de ABCD1 y normalizar la beta-oxidación, evitando la acumulación de AGCML); c) combinaciones de antioxidantes como la N-acetilcisteína con ácido lipoico y vitamina E (principalmente en AMN) o, d) administración de factores neurotróficos como el factor de crecimiento insulínico tipo 1 o la neurotropina 3.
En conclusión, si bien se precisan más estudios (controlados, amplios y duraderos) para confirmarlo, el aceite de Lorenzo podría ser eficaz para prevenir las manifestaciones neurológicas en pacientes asintomáticos con ADX-L. En pacientes con afectación neurológica, su empleo no modifica el curso de la enfermedad y el TPH realizado de forma precoz es actualmente el tratamiento de elección. En un futuro próximo, se espera que se desarrollen nuevas terapias.
