







Actualizar las recomendaciones sobre tratamiento antirretroviral (TAR) para adultos con infección por el VIH-1.
MétodosEste documento ha sido consensuado por un panel de expertos de GeSIDA y la Secretaría del Plan Nacional sobre el Sida tras revisar los resultados de eficacia y seguridad de ensayos clínicos, estudios de cohortes y de farmacocinética publicados en revistas biomédicas (PubMed y Embase) o presentados en congresos. La fuerza de cada recomendación y la gradación de su evidencia se basan en una modificación de los criterios de la Infectious Diseases Society of America.
ResultadosSe recomienda TAR a todos los pacientes con infección por VIH-1, aunque la fuerza y gradación de la recomendación varían en función del número de linfocitos CD4+/μl, la existencia de enfermedades oportunistas o comorbilidades, la edad y la prevención de la transmisión del VIH. El objetivo del TAR es lograr una carga viral plasmática (CVP) indetectable. El TAR inicial debe ser una combinación de 3 fármacos, que incluya 2 inhibidores de la transcriptasa inversa análogos de nucleósidos (ITIAN) y otro de distinta familia. Tres de las pautas recomendadas, todas las cuales tienen un inhibidor de la integrasa (INI) como tercer fármaco, se consideran preferentes, y otras 7, basadas en un INI, un inhibidor de la transcriptasa inversa no análogo de nucleósidos (ITINN) o un inhibidor de la proteasa potenciado con ritonavir (IP/r), como alternativas. Se exponen las causas y criterios para cambiar el TAR en los pacientes con CVP indetectable así como en los que presentan fracaso virológico, en cuyo caso el TAR de rescate debe incluir 3 (o al menos 2) fármacos plenamente activos frente al VIH. Se actualizan los criterios específicos del TAR en situaciones especiales (infección aguda, infección por VIH-2, embarazo) o comorbilidades (tuberculosis u otras enfermedades oportunistas, enfermedad renal, hepatopatías y neoplasias).
ConclusionesEste nuevo documento actualiza las recomendaciones previas respecto a cuándo y con qué regímenes iniciar el TAR, cómo monitorizarlo y qué hacer cuando fracasa o provoca toxicidad. Se actualizan los criterios específicos del TAR en pacientes con comorbilidades y en situaciones especiales.
This consensus document is an update of combined antiretroviral therapy (cART) guidelines and recommendations for HIV-1 infected adult patients.
MethodsTo formulate these recommendations, a panel composed of members of the AIDS Study Group and the AIDS National Plan (GeSIDA/Plan Nacional sobre el Sida) reviewed the efficacy and safety advances in clinical trials, and cohort and pharmacokinetic studies published in medical journals (PubMed and Embase) or presented in medical scientific meetings. The strength of the recommendations, and the evidence that supports them, are based on modified criteria of the Infectious Diseases Society of America.
ResultsIn this update, cART is recommended for all patients infected by type 1 human immunodeficiency virus (HIV-1). The strength and level of the recommendation depends on the CD4+T-lymphocyte count, the presence of opportunistic diseases or comorbid conditions, age, and prevention of transmission of HIV. The objective of cART is to achieve an undetectable plasma viral load. Initial cART should always comprise a combination of 3 drugs, including 2 nucleoside reverse transcriptase inhibitors, and a third drug from a different family. Three out of the ten recommended regimes are regarded as preferential (all of them with an integrase inhibitor as the third drug), and the other seven (based on a non-nucleoside reverse transcriptase inhibitor, a ritonavir-boosted protease inhibitor, or an integrase inhibitor) as alternatives. This update presents the causes and criteria for switching cART in patients with undetectable plasma viral load, and in cases of virological failure where rescue cART should comprise 3 (or at least 2) drugs that are fully active against the virus. An update is also provided for the specific criteria for cART in special situations (acute infection, HIV-2 infection, and pregnancy) and with comorbid conditions (tuberculosis or other opportunistic infections, kidney disease, liver disease, and cancer).
ConclusionsThese new guidelines update previous recommendations related to cART (when to begin and what drugs should be used), how to monitor and what to do in case of viral failure or drug adverse reactions. cART specific criteria in comorbid patients and special situations are equally updated.
Desde que se dispone de fármacos antirretrovirales (FAR) con los que formar combinaciones potentes, el tratamiento antirretroviral (TAR) ha logrado beneficios enormes en reducción de la morbimortalidad y transmisión de la infección por el VIH-1. Simultáneamente, el manejo de estos FAR ha adquirido gran complejidad con la aparición de distintas familias y de múltiples facetas en cuanto a eficacia, toxicidad, resistencias, tropismo, interacciones, uso en situaciones clínicas especiales y como prevención de la transmisión del VIH-1, estudios de coste-eficacia, etc. Por esta complejidad, y por la rapidez con que se incrementan los conocimientos, se exige no solo la elaboración de guías y recomendaciones sobre TAR, sino también su actualización frecuente. A este respecto, el Grupo de Estudio de Sida (GeSIDA) de la Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC) y el Plan Nacional sobre el Sida (PNS) editan conjuntamente desde hace 16 años un documento de consenso sobre TAR en adultos que, al igual que los de otras instituciones y sociedades científicas1-3, se ha venido actualizando anualmente. Dichas actualizaciones se publican tanto en la revista Enfermedades Infecciosas y Microbiología Clínica como en las páginas de GeSIDA (www.gesida-seimc.org) y del PNS (www.msssi.gob.es/ciudadanos/enfLesiones/enfTransmisibles/sida/planNalSida).
En estas recomendaciones no se detallan determinados aspectos del TAR (adherencia, uso en pacientes coinfectados por los virus de las hepatitis B o C, insuficiencia renal, tratamiento antituberculosos simultáneo, aspectos relativos a la mujer —deseo reproductivo, embarazo, parto, profilaxis de la transmisión vertical y menopausia—, profilaxis postexposición, etc.) que se han elaborado en colaboración con otras sociedades científicas y a cuyas publicaciones se remite al lector interesado.
Los objetivos de este documento de consenso son facilitar a los profesionales que tratan a adultos con infección por el VIH-1 el estado actual del conocimiento sobre el TAR y proporcionarles recomendaciones basadas en evidencias científicas que puedan guiar sus decisiones terapéuticas.
MetodologíaEl Panel redactor del documento está integrado por clínicos expertos en la infección por el VIH-1 y el TAR designados por la Junta Directiva de GeSIDA y el PNS, que han aceptado participar voluntariamente y emitir una declaración de conflicto de intereses. Estos expertos se distribuyen en grupos formados por un redactor y varios consultores que se encargan de actualizar una sección del documento. Tres miembros del Panel (presidente y secretario de GeSIDA y la responsable del Área Asistencial del PNS) actúan como coordinadores y otros 2 como redactores generales, cuyo cometido ha sido ensamblar todas las secciones del documento y encargarse de la redacción y edición final del mismo. El redactor de cada grupo revisó los datos más relevantes de las publicaciones científicas (PubMed y Embase; idiomas: español, inglés y francés) y de las comunicaciones a los congresos más recientes hasta el 30 de octubre de 2014.
El texto elaborado por el redactor se somete a la consideración de sus consultores y se incorporan las aportaciones aceptadas por consenso. Una vez ensambladas todas las secciones, el documento se discute y consensúa en una reunión presencial del Panel. Tras la incorporación de las modificaciones aprobadas en dicha reunión, el documento se expone durante al menos 15 días en las páginas web de GeSIDA y del PNS para que los profesionales, los pacientes o quien esté interesado pueda hacer sugerencias que, tras su estudio y deliberación, pueden ser integradas en el documento final. El Panel considera que, en el caso de aparecer nuevas evidencias relevantes que impliquen cambios en las recomendaciones sobre TAR, serán incorporadas al documento en las páginas web y, a ser posible, en la revista.
En este documento la fuerza de la recomendación y gradación de las pruebas que la sustentan se basan en una modificación de los criterios de la Infectious Diseases Society of America4, según los cuales cada recomendación debe ofrecerse siempre (A), en general (B) u opcionalmente (C), y ello basado en los resultados obtenidos a partir de uno o más ensayos clínicos aleatorizados de aspectos clínicos o de laboratorio (I), de uno o más ensayos no aleatorizados o datos observacionales de cohortes (II), o en la opinión de expertos (III).
GeSIDA y el PNS seguirán actualizando este documento periódicamente en función de la evolución de los conocimientos sobre el TAR. No obstante se recuerda que, dado que estos cambian muy rápidamente, es conveniente que los lectores consulten también otras fuentes de información.
Para el control del seguimiento y la evaluación del impacto de estas recomendaciones sobre la respuesta al TAR de los pacientes seguidos en cada centro o profesional, pueden utilizarse algunos de los parámetros específicos incluidos en el documento de indicadores de calidad asistencial en la atención a personas infectadas por el VIH-1, elaborado por GeSIDA5.
Evaluación clínica y de laboratorio para guiar el tratamiento antirretroviralEvaluación clínicaSe realizará una historia clínica completa incluyendo una evaluación psicológica y psiquiátrica, riesgo cardiovascular, vacunaciones, viajes, hábitos sexuales, salud sexual de la pareja, uso de drogas y tóxicos, y una evaluación farmacológica exhaustiva para prevenir o detectar posibles interacciones entre los fármacos utilizados para tratar las comorbilidades y el TAR6,7. En las mujeres, además, es conveniente investigar aspectos relacionados con el deseo reproductivo, anticoncepción y otros aspectos ginecológicos específicos. También se debe realizar un examen físico general que incluya piel, orofaringe, corazón, pulmón, abdomen, ganglios linfáticos periféricos, musculoesquelético, neurológico con evaluación neurocognitiva, y anogenital. Esta evaluación se repetirá con periodicidad anual y/o siempre que la situación clínica del paciente lo requiera6,7.
Recomendación- –
Se debe realizar historia clínica, evaluación farmacológica y examen físico a todo paciente infectado por el VIH-1, que se repetirá con periodicidad anual (A-II).
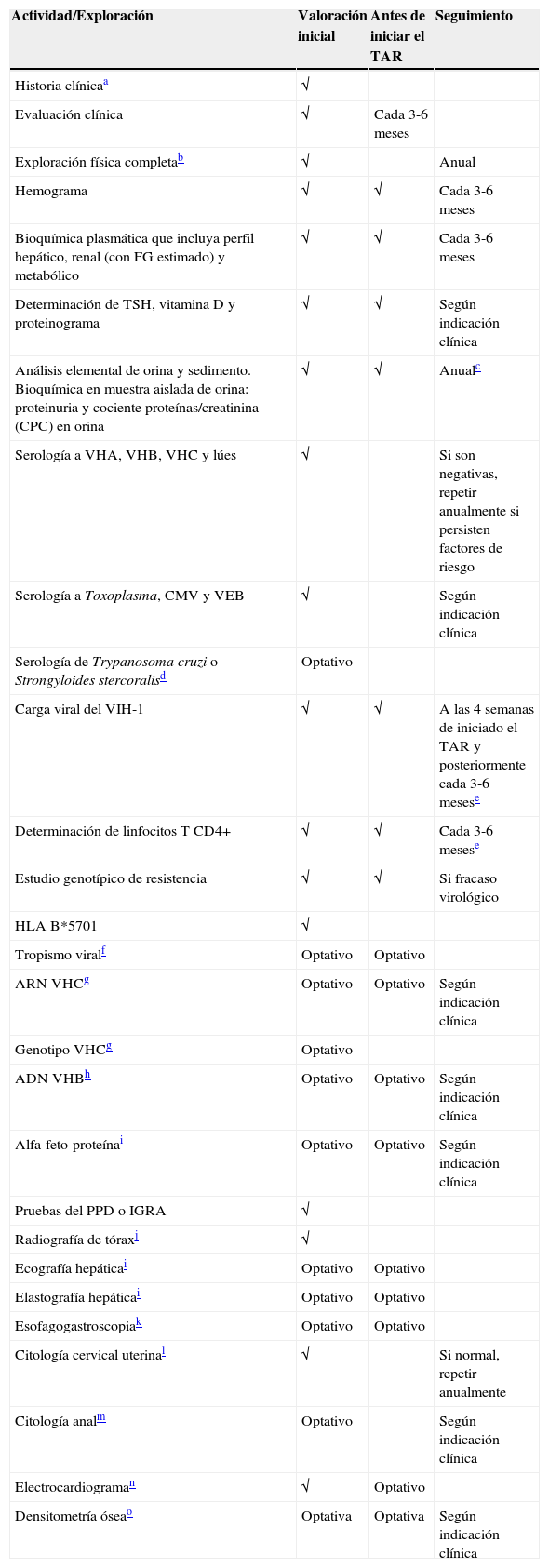
Se efectuarán determinaciones analíticas generales y, además, otras determinaciones específicas relativas al VIH-1. Los análisis generales incluyen hemograma, bioquímica básica con determinación de creatinina, filtrado glomerular estimado (FGe, preferiblemente mediante las ecuaciones CKD-EPI o MDRD), sodio, potasio, enzimas hepáticas, perfil metabólico (glucosa, colesterol total, colesterol HDL, colesterol LDL y triglicéridos), TSH, proteinograma y perfil óseo (calcio, fósforo y vitamina D), análisis elemental y sedimento de orina, con cuantificación de proteinuria y cálculo del cociente proteína/creatinina en orina (con diabetes mellitus o hipertensión arterial, cuantificar también la albuminuria y calcular el cociente albúmina/creatinina en orina). Se debe efectuar una serología de toxoplasma, citomegalovirus, lúes, VHA, VHB y VHC. En relación a la infección por el propio VIH-1, se efectuará un recuento de linfocitos CD4+, carga viral plasmática (CVP), estudio de resistencias virales, determinación de HLA-B*5701 y, eventualmente, estudio del tropismo viral. El recuento de linfocitos CD4+ y la CVP son los parámetros que se utilizan, además de la evaluación clínica, para indicar el TAR, monitorizar su efectividad y tomar decisiones respecto a cambios del tratamiento6,7. En la tabla 1 se describen los estudios complementarios recomendados en la evaluación inicial y el seguimiento periódico de los pacientes con infección por el VIH-1. A continuación se comenta con mayor detalle la información relacionada con las determinaciones específicamente relacionadas con el VIH-1.
Exploraciones complementarias en la valoración inicial y el seguimiento de los pacientes con infección por el VIH-1
| Actividad/Exploración | Valoración inicial | Antes de iniciar el TAR | Seguimiento |
|---|---|---|---|
| Historia clínicaa | √ | ||
| Evaluación clínica | √ | Cada 3-6 meses | |
| Exploración física completab | √ | Anual | |
| Hemograma | √ | √ | Cada 3-6 meses |
| Bioquímica plasmática que incluya perfil hepático, renal (con FG estimado) y metabólico | √ | √ | Cada 3-6 meses |
| Determinación de TSH, vitamina D y proteinograma | √ | √ | Según indicación clínica |
| Análisis elemental de orina y sedimento. Bioquímica en muestra aislada de orina: proteinuria y cociente proteínas/creatinina (CPC) en orina | √ | √ | Anualc |
| Serología a VHA, VHB, VHC y lúes | √ | Si son negativas, repetir anualmente si persisten factores de riesgo | |
| Serología a Toxoplasma, CMV y VEB | √ | Según indicación clínica | |
| Serología de Trypanosoma cruzi o Strongyloides stercoralisd | Optativo | ||
| Carga viral del VIH-1 | √ | √ | A las 4 semanas de iniciado el TAR y posteriormente cada 3-6 mesese |
| Determinación de linfocitos T CD4+ | √ | √ | Cada 3-6 mesese |
| Estudio genotípico de resistencia | √ | √ | Si fracaso virológico |
| HLA B*5701 | √ | ||
| Tropismo viralf | Optativo | Optativo | |
| ARN VHCg | Optativo | Optativo | Según indicación clínica |
| Genotipo VHCg | Optativo | ||
| ADN VHBh | Optativo | Optativo | Según indicación clínica |
| Alfa-feto-proteínai | Optativo | Optativo | Según indicación clínica |
| Pruebas del PPD o IGRA | √ | ||
| Radiografía de tóraxj | √ | ||
| Ecografía hepáticai | Optativo | Optativo | |
| Elastografía hepáticai | Optativo | Optativo | |
| Esofagogastroscopiak | Optativo | Optativo | |
| Citología cervical uterinal | √ | Si normal, repetir anualmente | |
| Citología analm | Optativo | Según indicación clínica | |
| Electrocardiograman | √ | Optativo | |
| Densitometría óseao | Optativa | Optativa | Según indicación clínica |
Estas recomendaciones deben considerarse orientativas y pueden ser modificadas de acuerdo con el juicio clínico de los profesionales responsables de la atención al paciente.
√ Realizar
Si se usa TDF, realizar cada 3-4 meses. Si diabetes mellitus o hipertensión arterial, determinar además albuminuria y cociente albúmina/creatinina (CAC) en muestra aislada de orina.
Optativo en personas procedentes de áreas con alta prevalencia de infestación, sobre todo si se sospecha la misma (p. ej., eosinofilia).
Se puede considerar determinar la CVP y los linfocitos T CD4+ con menos frecuencia (cada 6-12 meses) en pacientes clínicamente estables, con CVP repetidamente suprimida y cifras de linfocitos T CD4+ repetidamente superiores a 500 células/μl.
Si coinfección por VHC, VHB u otra causa de daño hepático crónico, periodicidad según indicación clínica (cada 6-12 meses).
Especialmente en pacientes pertenecientes a poblaciones con una elevada prevalencia de tuberculosis.
Si cirrosis hepática confirmada o elastografía >12,5kPpa (F4), para descartar varices; periodicidad según indicación clínica.
Debe realizarse inicialmente en todos los pacientes con hábitos de riesgo (varones homo/bisexuales y mujeres que practican coito anal receptivo) y en los que presentan lesiones perianales o genitales secundarias a VPH. Si citología anormal, debe realizarse anuscopia de alta resolución y biopsia (evidencia de beneficio desconocida; defendida por algunos expertos).
- –
La evaluación inicial de laboratorio debe incluir: hemograma, bioquímica general, serología de toxoplasma, citomegalovirus, lúes, VHA, VHB y VHC; CVP, linfocitos CD4+, mutaciones de resistencia (MR) primarias y HLA-B*5701 (A-II).
La cifra de linfocitos CD4+ es el principal indicador del estado inmunológico. Usualmente se utiliza el recuento absoluto, pero también puede utilizarse su porcentaje, que es más estable y objetivo, particularmente en pacientes con leucopenia. Se utiliza para estadificar la infección por VIH-1 y evaluar la vulnerabilidad a determinadas infecciones oportunistas, la necesidad de su profilaxis primaria7 y la eventual discontinuación8. Hoy día se reconoce la indicación universal de TAR a todos los pacientes, independientemente del recuento de linfocitos CD4+, aunque en función de este valor la fortaleza de la evidencia para justificar la indicación del TAR varía (de A-I a B-III). Una vez instaurado este, el aumento de linfocitos CD4+, en los pacientes en que están disminuidos, suele ser lento pero constante en el tiempo. Se admite que durante el primer año debería existir un aumento mínimo de 50-100 linfocitos CD4+/μl9, aunque en algunos pacientes muy inmunodeprimidos no se produce a pesar de la supresión virológica.
En los pacientes asintomáticos que no reciben TAR los linfocitos CD4+ deben medirse cada 3-6 meses y repetirse en 4 semanas ante un hallazgo que justifique tomar una decisión terapéutica10. Una vez iniciado el TAR, se determinarán a las 4 semanas y posteriormente cada 3-6 meses o siempre que cambios en la situación clínica lo hagan aconsejable. Los controles pueden ser más espaciados (hasta 12 meses) en los pacientes clínicamente estables, con CVP suprimida y cifras de linfocitos CD4+ repetidamente mayores de 300-500 células/μl11,12.
La monitorización debe ser más frecuente en las situaciones clínicas que pueden disminuirlos (tratamiento con interferón, fármacos antineoplásicos, uso de corticoides, etc.) por riesgo de desarrollo de infecciones oportunistas.
Recomendaciones- –
Se debe determinar periódicamente el número absoluto y el porcentaje de linfocitos CD4+ antes de iniciar el TAR y, una vez iniciado, como parámetro de monitorización periódica de la respuesta inmunológica al mismo (A-I).
- –
Los controles pueden ser más espaciados (hasta 12 meses) en los pacientes estables, con CVP suprimida y cifras de linfocitos por encima de 300-500 células/μl (C-II).
La CVP se ha de determinar antes de iniciar el TAR, ya que desciende rápidamente tras su inicio7,13. El objetivo de supresión de la CVP es conseguir unas cifras inferiores a 20-50 copias/ml, ya que así no se seleccionan MR14 y la duración de la respuesta virológica es mayor que con cifras entre 50-500 copias/ml. Los pacientes con CVP muy elevadas pueden tardar hasta 24 semanas en conseguir niveles inferiores a 20-50 copias/ml. En pacientes con CVP habitualmente indetectable no es infrecuente detectar viremia de bajo nivel (blips), que vuelve espontáneamente a ser indetectable sin ningún cambio en el TAR. Aunque en la mayoría de estudios no se ha observado que los blips aumenten el riesgo de fracaso virológico (FV), en algunos pacientes pueden seleccionar MR15.
Se entiende por respuesta virológica la reducción de la CVP en más de 1 log a las 4 semanas del inicio del TAR y ser indetectable (<50 copias/ml) tras 16-24 semanas de tratamiento. Por el contrario, hablamos de fracaso virológico (FV) si se dan cualesquiera de las 2 situaciones siguientes: a)CVP detectable tras 24 semanas del inicio del TAR, y b)si, tras alcanzar la indetectabilidad, la CVP vuelve a ser >50 copias/ml en 2 determinaciones consecutivas (separadas por 2-4 semanas).
Es conveniente medir la CVP a las 4 semanas del inicio del TAR, y posteriormente cada 3-6 meses, para comprobar la respuesta virológica y como medida indirecta de adherencia al TAR y refuerzo de la misma16. En pacientes clínicamente estables con CVP repetidamente suprimida y cifras elevadas de linfocitos CD4+ este intervalo de tiempo puede alargarse incluso hasta 12 meses17. Si la medida de la CVP se efectúa tras un proceso infeccioso intercurrente o vacunación, puede haber elevaciones transitorias.
Recomendaciones- –
Se debe determinar la CVP antes del inicio del TAR (A-II).
- –
La CVP es el parámetro principal para evaluar la eficacia virológica del TAR y para definir el FV (A-I).
- –
Los objetivos de supresión virológica se deben conseguir tanto en pacientes sin TAR previo como en individuos que han experimentado un fracaso previo (A-II).
- –
Debe utilizarse una técnica de determinación de CVP con un límite de cuantificación de al menos 50 copias/ml y usar siempre la misma técnica (A-II).
- –
Si se van a tomar decisiones terapéuticas en función de un resultado de la CVP, se debe confirmar con una segunda determinación (A-II).
Aunque sexo, edad, peso, superficie corporal, determinantes genéticos del huésped, interacciones medicamentosas, embarazo e insuficiencia hepática o renal se han asociado a variaciones importantes en los niveles plasmáticos de FAR18, son múltiples las limitaciones para su uso rutinario en la clínica diaria: resultados no concordantes de su eficacia en estudios prospectivos19, desconocimiento de rangos terapéuticos asociados a respuesta o reducción de efectos adversos, variabilidad intraindividual, no disponibilidad para los inhibidores de la transcriptasa inversa análogos de nucleósidos (ITIAN) y falta de disponibilidad de la técnica en los laboratorios.
Recomendaciones- –
No se recomienda la medición de concentraciones plasmáticas de FAR para el control habitual del paciente con infección por el VIH-1 (A-II).
- –
La medición de concentraciones plasmáticas de FAR puede estar indicada en algunas situaciones clínicas: riesgo de interacciones farmacológicas, trasplantes de órganos, delgadez extrema u obesidad mórbida, embarazo, insuficiencia hepática o renal, etc., así como para confirmar la sospecha de un cumplimiento terapéutico deficiente (B-III).
El VIH-1 tiene una gran tendencia a producir mutantes resistentes. Las cuasiespecies de VIH-1 con MR en general tienen menor capacidad replicativa (fitness)20. Las resistencias pueden investigarse mediante técnicas genotípicas, que detectan cambios específicos en el genoma de las enzimas diana de los FAR (transcriptasa inversa, proteasa, integrasa) o fenotípicas, que determinan la respuesta de la población viral mayoritaria a concentraciones crecientes de los FAR20. Ambas comparten limitaciones en especificidad (no detectan poblaciones por debajo del 20%) y sensibilidad (CVP<500-1.000 copias/ml)21, aunque existen técnicas que soslayan estos inconvenientes. Las técnicas genotípicas son las que se utilizan en la asistencia clínica, dado que son más sencillas, rápidas y accesibles.
En los pacientes con FV es imperativo efectuar un estudio de resistencias para adecuar el nuevo TAR al perfil de mutaciones del VIH-120,21, incluyendo resistencias en la integrasa viral si el paciente ha fracasado a FAR de esta familia. Debido a la capacidad de reversión de las mutaciones a las cepas sensibles, es necesario efectuar el estudio manteniendo el TAR activo.
Los pacientes con nuevo diagnóstico pueden haberse infectado por cepas de VIH-1 resistentes a algunos FAR (resistencia primaria), por lo que es necesario efectuar un estudio de resistencias en el momento del diagnóstico. Sin embargo, las cepas con mutaciones pueden revertir o convertirse en minoritarias a los pocos meses (menor fitness). Si se difiere el inicio del TAR en un paciente recientemente diagnosticado, es conveniente un nuevo estudio de resistencias antes de iniciarlo22.
La transmisión de resistencias primarias en países occidentales se ha estabilizado23, y la última actualización en la cohorte de la Red de SIDA (CoRIS) para el periodo 2013-2014 ha estimado una prevalencia global de resistencias primarias a inhibidores de la transcriptasa inversa y de la proteasa del 8,7%24. La transmisión de resistencias en la integrasa es excepcional25, por lo que su determinación en pacientes sin TAR previo se reserva solo para aquellos en los que existe alta sospecha de transmisión (multirresistencia en proteasa y/o transcriptasa inversa o caso índice tratado con inhibidores de la integrasa [INI]).
En relación a las variantes minoritarias, la información disponible sugiere que pueden estar implicadas en algunos fracasos virológicos26, y cada vez se les concede un mayor interés, especialmente si el TAR se inicia con inhibidores de la transcriptasa inversa noanálogos de los nucleósidos (ITINN).
Para la interpretación de las mutaciones asociadas con resistencia a FAR se utilizan algoritmos que se actualizan periódicamente, como el de la CoRIS (www.retic-ris.net).
Recomendaciones- –
Se recomienda realizar un estudio genotípico de resistencias del VIH-1 en los genes de la transcriptasa inversa y proteasa en todos los pacientes, tanto al diagnóstico de la infección como antes de iniciar el TAR si se difiere (A-II).
- –
Solo se recomienda estudiar resistencias en la integrasa si existe alta sospecha de transmisión de resistencias a esta familia (C-III).
- –
Se recomienda la realización de un estudio genotípico de resistencias del VIH-1 en todos los pacientes con FV confirmado (A-I).
Los portadores del alelo HLA-B*5701 tienen mayor riesgo de presentar una reacción de hipersensibilidad (RHS) a abacavir (ABC). La RHS a ABC es un síndrome multiorgánico que puede manifestarse con una combinación variable de exantema, fiebre, mialgias y síntomas respiratorios o gastrointestinales que puede llegar a ser fatal si se continúa tomando ABC o se reintroduce este FAR después de haberlo suspendido. Suele aparecer durante las primeras 6 semanas de TAR y se presenta en el 5-8% de los pacientes que toman ABC27. Ensayos clínicos en diferentes poblaciones (caucásica y raza negra, en la que la prevalencia de este alelo es menor) han demostrado fehacientemente que cuando no se prescribe ABC a los pacientes que son portadores de HLA-B*5701, disminuye significativamente la incidencia de RHS28,29. ABC no debe utilizarse, por tanto, en personas portadoras de HLA-B*5701. Una prueba de HLA-B*5701 negativa no descarta completamente la posibilidad de RHS por ABC, por lo cual se debe informar a los pacientes acerca de la RHS cuando se inicia un tratamiento con ABC y evaluar los síntomas que eventualmente puedan presentar.
Recomendaciones- –
Se debe determinar el HLA-B*5701 en todos los pacientes antes de iniciar un régimen de TAR que contenga ABC (A-I).
- –
No se debe prescribir ABC si la prueba del HLA-B*5701 es positiva (A-I).
La disponibilidad de maraviroc (MVC), FAR antagonista del correceptor CCR5, requiere conocer el tropismo del VIH-1 para ser usado. MVC se debe prescribir exclusivamente en los pacientes con infección por cepas R5 de VIH-1. El tropismo del VIH se determina mediante métodos genotípicos, que determinan la secuencia de la región V3 de la gp120. Esta es la técnica de referencia actual, ya que se puede efectuar en cualquier laboratorio que realice estudios de resistencias, es rápida, no precisa una CVP elevada para obtener resultados fiables y reproducibles y su coste es aceptable30. Existen métodos fenotípicos, como el TrofileESTA®, pero son técnicas complejas y caras.
Diferentes grupos de consenso nacionales31 y europeos32 recomiendan determinar el tropismo en los pacientes que hayan fracasado a cualquier línea de tratamiento y vayan a iniciar un tratamiento de rescate que contemple el uso de antagonistas de CCR5, y en pacientes sin TAR previo en las que un FAR antagonista del receptor CCR5 pueda considerarse una buena opción terapéutica.
Recomendación- –
Se debe determinar el tropismo viral antes de iniciar el tratamiento con un FAR inhibidor del receptor CCR5 (A-I).
Los principales motivos para iniciar el TAR son la reducción de la morbimortalidad asociada a la infección por VIH, la recuperación y preservación de la función inmunológica, evitar el efecto nocivo de la replicación del VIH-1 sobre posibles comorbilidades existentes y la prevención de la transmisión del VIH. Es importante valorar de forma individual el momento de inicio del TAR y de los FAR que deben formar parte del régimen inicial, sopesando las ventajas e inconvenientes de cada una de las opciones. La disposición y la motivación del paciente es un factor crítico a la hora de tomar la decisión de cuándo empezarlo.
Cuándo iniciar el tratamiento antirretroviralNo existe ninguna duda sobre la necesidad de tratar a todos los pacientes con sintomatología relacionada con la infección por el VIH-1 (eventos clínicos B o C de la clasificación de los CDC de 2003, incluyendo la nefropatía por VIH), puesto que el tratamiento se relaciona con mejora de la supervivencia.
En pacientes asintomáticos, los resultados de diferentes ensayos clínicos indican que el riesgo de progresión y/o muerte es mayor cuando se inicia el TAR con cifras de linfocitos CD4+ inferiores a 350 células/μl que cuando se inicia con cifras por encima de esta33,34. La mejora en la seguridad de los tratamientos y la acumulación de datos que indican una mayor morbimortalidad por causas no relacionadas con el sida en pacientes asintomáticos con menos de 350 linfocitos CD4+/μl en diferentes estudios observacionales y de subanálisis de algunos ensayos clínicos33,35 han modificado la recomendación de inicio del TAR en este grupo de pacientes.
En la ART Cohort Collaboration36 el riesgo de progresión y/o muerte fue mayor cuando se inició el TAR entre 250-350 linfocitos CD4+/μl que con 350-450/μl; sin embargo, el inicio con cifras de linfocitos CD4+ entre 450-550/μl no se relacionó con un descenso adicional del riesgo de progresión o muerte. En la cohorte HIV-CAUSAL37 el riesgo de progresión a sida o muerte fue mayor cuando se inició el TAR con menos de 350 linfocitos CD4+/μl que cuando se inició entre 350 y 500/μl. En la CASCADE Collaboration38, iniciar TAR entre 350 y 500 linfocitos CD4+/μl también disminuyó la mortalidad en comparación con la de los pacientes que lo iniciaron con cifras de CD4+ inferiores a 350/μl; sin embargo, empezar TAR entre 500 y 799 CD4+/μl no disminuyó el riesgo de progresión a sida o muerte con respecto al grupo que lo inició entre 350 y 500 CD4+/μl. Por el contrario, los datos de la cohorte NA-ACCORD39 muestran un mayor riesgo de muerte en los pacientes en los que se demora el inicio del TAR hasta recuentos de linfocitos CD4+ inferiores a 500/μl, comparado con los que lo inician más precozmente.
En un pequeño subestudio del ensayo SMART34, los pacientes que iniciaron TAR con cifras de linfocitos CD4+ mayores de 350/μl (mediana: 437/μl) presentaron menos complicaciones graves asociadas o no al sida que los que lo hicieron con menos de 250 células/μl. Por último, en el estudio HPTN 05235, iniciar TAR entre 350-550 CD4+/μl disminuyó el riesgo de progresión clínica, pero no la mortalidad, con respecto a los que lo iniciaron con menos de 250 CD4+/μl.
Todos estos datos favorecen la recomendación de iniciar TAR con menos de 500 CD4+/μl. El debate actual se centra en torno a si es necesario iniciar TAR con más de 500 linfocitos CD4+/μl. No existen de momento datos concluyentes, aunque la simplicidad y la tolerabilidad de los regímenes actuales tampoco justifican la demora para iniciar el TAR. Además, el incremento de la población tratada se ha relacionado con una disminución de las tasas de transmisión y, por tanto, de la disminución de nuevas infecciones40,41. Por todo ello, la recomendación de iniciar el TAR en toda persona con infección por el VIH-1 es compartida por un número creciente de expertos. En cualquier caso, el TAR debería recomendarse en aquellos grupos de pacientes con un mayor riesgo de progresión, como los que presentan CVP superior a 105 copias/ml y una edad igual o mayor de 55años. También se recomienda iniciar el TAR en pacientes con comorbilidades cuya progresión se ve facilitada por la propia infección por VIH, como la cirrosis hepática, la hepatitis crónica por VHC, la existencia de riesgo cardiovascular elevado, los tumores no relacionados con el sida y los trastornos neurocognitivos.
Por último, existen determinadas circunstancias en las que el TAR debe recomendarse con independencia de la situación inmunológica, como es el caso de las mujeres embarazadas (para disminuir el riesgo de transmisión maternofetal), la coinfección por hepatitis B subsidiaria de tratamiento antiviral, o las parejas serodiscordantes que deseen disminuir al máximo el riesgo de transmisión del VIH.
Como excepción se consideran los pacientes que mantienen CVP indetectable de forma mantenida sin TAR (controladores de élite). En este caso no existe información que permita valorar el efecto beneficioso del TAR, por lo que no se puede establecer una recomendación al respecto.
Recomendaciones- –
Se recomienda la administración de TAR a todos los pacientes con infección por el VIH-1 para evitar la progresión de la enfermedad, disminuir la transmisión del virus y limitar el efecto nocivo sobre posibles comorbilidades coexistentes. La fuerza de la recomendación varía según las circunstancias, como se detalla en la tabla 2.
Tabla 2.Indicaciones de tratamiento antirretroviral en pacientes con infección crónica por el VIHa
Recomendación general Se recomienda la administración de TAR a todos los pacientes con infección por VIHb. La fuerza y gradación de la recomendación varía según las siguientes circunstancias: Condición/circunstancia Fuerza y gradación Enfermedades B o C del CDC A-I Cifra de linfocitos T CD4+ < 350/μl A-I 350 a 500/μl A-II > 500/μl B-III Comorbilidades A-II Nefropatía por VIH Hepatitis crónica por VHC Hepatitis crónica por VHB Edad ≥ 55 años Riesgo cardiovascular elevado Trastornos neurocognitivos Neoplasias Riesgo de transmisión Mujeres gestantes A-I Transmisión heterosexual A-I Transmisión sexual entre varones A-III aEs importante hacer una valoración individualizada del momento de inicio del TAR y de los FAR que deben formar parte del régimen inicial, sopesando las ventajas e inconvenientes de cada una de las opciones. La disposición y la motivación del paciente es un factor crítico a la hora de tomar la decisión de cuándo empezarlo.
- –
El inicio del TAR debe valorarse siempre individualmente. Antes de tomar la decisión de iniciarlo deben confirmarse las cifras de linfocitos CD4+ y CVP. Además, debe prepararse al paciente, ofertando las distintas opciones, adaptando el esquema terapéutico al estilo de vida, comorbilidades y posibles interacciones y valorando el riesgo de mala adherencia (A-III).
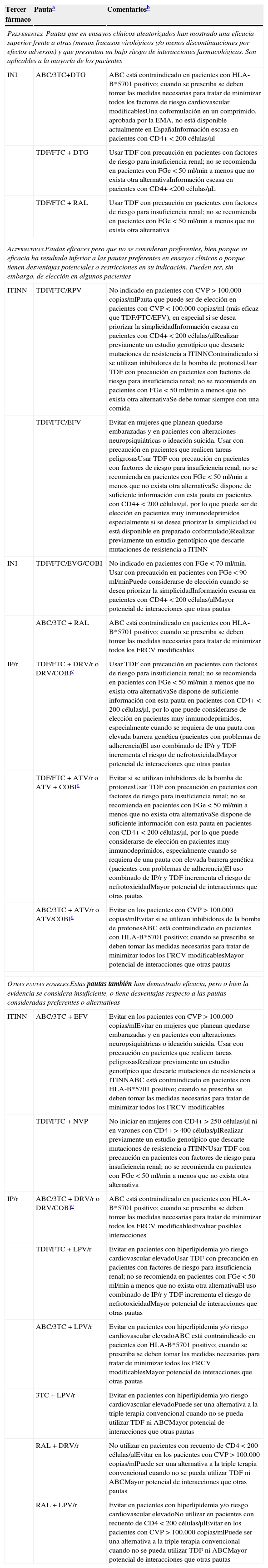
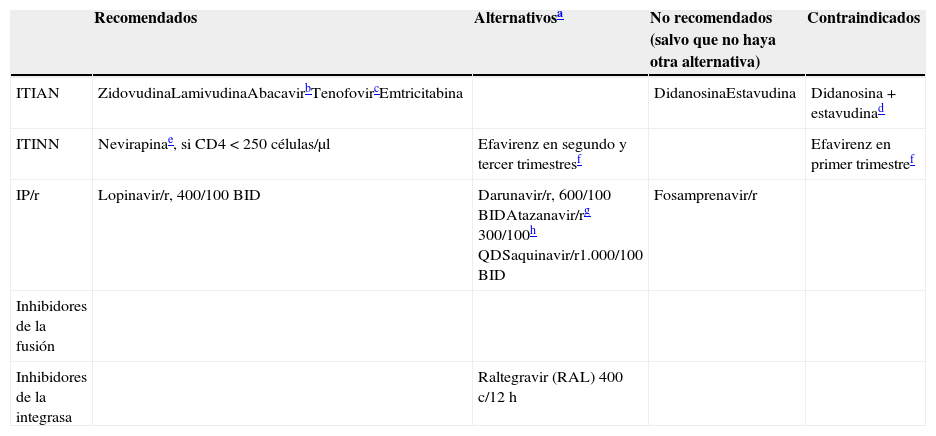
Las pautas recomendadas para el tratamiento inicial de la infección por el VIH-1 en el momento actual consisten en una combinación de 3 fármacos que incluyan 2 ITIAN asociados a un INI, un ITINN, o un inhibidor de la proteasa (IP) potenciado con ritonavir (IP/r) (tabla 3). Con estas combinaciones se puede conseguir una CVP inferior a 50 copias/ml en más del 75% de los casos a las 48 semanas.
Combinaciones de TAR de inicio recomendadasa
| Tercer fármaco | Pautaa | Comentariosb |
|---|---|---|
| Preferentes. Pautas que en ensayos clínicos aleatorizados han mostrado una eficacia superior frente a otras (menos fracasos virológicos y/o menos discontinuaciones por efectos adversos) y que presentan un bajo riesgo de interacciones farmacológicas. Son aplicables a la mayoría de los pacientes | ||
| INI | ABC/3TC+DTG | ABC está contraindicado en pacientes con HLA-B*5701 positivo; cuando se prescriba se deben tomar las medidas necesarias para tratar de minimizar todos los factores de riesgo cardiovascular modificablesUna coformulación en un comprimido, aprobada por la EMA, no está disponible actualmente en EspañaInformación escasa en pacientes con CD4+ < 200 células/μl |
| TDF/FTC + DTG | Usar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativaInformación escasa en pacientes con CD4+ <200 células/μL | |
| TDF/FTC + RAL | Usar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativa | |
| Alternativas.Pautas eficaces pero que no se consideran preferentes, bien porque su eficacia ha resultado inferior a las pautas preferentes en ensayos clínicos o porque tienen desventajas potenciales o restricciones en su indicación. Pueden ser, sin embargo, de elección en algunos pacientes | ||
| ITINN | TDF/FTC/RPV | No indicado en pacientes con CVP > 100.000 copias/mlPauta que puede ser de elección en pacientes con CVP < 100.000 copias/ml (más eficaz que TDF/FTC/EFV), en especial si se desea priorizar la simplicidadInformación escasa en pacientes con CD4+ < 200 células/μlRealizar previamente un estudio genotípico que descarte mutaciones de resistencia a ITINNContraindicado si se utilizan inhibidores de la bomba de protonesUsar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativaSe debe tomar siempre con una comida |
| TDF/FTC/EFV | Evitar en mujeres que planean quedarse embarazadas y en pacientes con alteraciones neuropsiquiátricas o ideación suicida. Usar con precaución en pacientes que realicen tareas peligrosasUsar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativaSe dispone de suficiente información con esta pauta en pacientes con CD4+ < 200 células/μl, por lo que puede ser de elección en pacientes muy inmunodeprimidos especialmente si se desea priorizar la simplicidad (si está disponible en preparado coformulado)Realizar previamente un estudio genotípico que descarte mutaciones de resistencia a ITINN | |
| INI | TDF/FTC/EVG/COBI | No indicado en pacientes con FGe < 70 ml/min. Usar con precaución en pacientes con FGe < 90 ml/minPuede considerarse de elección cuando se desea priorizar la simplicidadInformación escasa en pacientes con CD4+ < 200 células/μlMayor potencial de interacciones que otras pautas |
| ABC/3TC + RAL | ABC está contraindicado en pacientes con HLA-B*5701 positivo; cuando se prescriba se deben tomar las medidas necesarias para tratar de minimizar todos los FRCV modificables | |
| IP/r | TDF/FTC + DRV/r o DRV/COBIc | Usar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativaSe dispone de suficiente información con esta pauta en pacientes con CD4+ < 200 células/μl, por lo que puede considerarse de elección en pacientes muy inmunodeprimidos, especialmente cuando se requiera de una pauta con elevada barrera genética (pacientes con problemas de adherencia)El uso combinado de IP/r y TDF incrementa el riesgo de nefrotoxicidadMayor potencial de interacciones que otras pautas |
| TDF/FTC + ATV/r o ATV + COBIc | Evitar si se utilizan inhibidores de la bomba de protonesUsar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativaSe dispone de suficiente información con esta pauta en pacientes con CD4+ < 200 células/μl, por lo que puede considerarse de elección en pacientes muy inmunodeprimidos, especialmente cuando se requiera de una pauta con elevada barrera genética (pacientes con problemas de adherencia)El uso combinado de IP/r y TDF incrementa el riesgo de nefrotoxicidadMayor potencial de interacciones que otras pautas | |
| ABC/3TC + ATV/r o ATV/COBIc | Evitar en los pacientes con CVP > 100.000 copias/mlEvitar si se utilizan inhibidores de la bomba de protonesABC está contraindicado en pacientes con HLA-B*5701 positivo; cuando se prescriba se deben tomar las medidas necesarias para tratar de minimizar todos los FRCV modificablesMayor potencial de interacciones que otras pautas | |
| Otras pautas posibles.Estas pautas también han demostrado eficacia, pero o bien la evidencia se considera insuficiente, o tiene desventajas respecto a las pautas consideradas preferentes o alternativas | ||
| ITINN | ABC/3TC + EFV | Evitar en los pacientes con CVP > 100.000 copias/mlEvitar en mujeres que planean quedarse embarazadas y en pacientes con alteraciones neuropsiquiátricas o ideación suicida. Usar con precaución en pacientes que realicen tareas peligrosasRealizar previamente un estudio genotípico que descarte mutaciones de resistencia a ITINNABC está contraindicado en pacientes con HLA-B*5701 positivo; cuando se prescriba se deben tomar las medidas necesarias para tratar de minimizar todos los FRCV modificables |
| TDF/FTC + NVP | No iniciar en mujeres con CD4+ > 250 células/μl ni en varones con CD4+ > 400 células/μlRealizar previamente un estudio genotípico que descarte mutaciones de resistencia a ITINNUsar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativa | |
| IP/r | ABC/3TC + DRV/r o DRV/COBIc | ABC está contraindicado en pacientes con HLA-B*5701 positivo; cuando se prescriba se deben tomar las medidas necesarias para tratar de minimizar todos los FRCV modificablesEvaluar posibles interacciones |
| TDF/FTC + LPV/r | Evitar en pacientes con hiperlipidemia y/o riesgo cardiovascular elevadoUsar TDF con precaución en pacientes con factores de riesgo para insuficiencia renal; no se recomienda en pacientes con FGe < 50 ml/min a menos que no exista otra alternativaEl uso combinado de IP/r y TDF incrementa el riesgo de nefrotoxicidadMayor potencial de interacciones que otras pautas | |
| ABC/3TC + LPV/r | Evitar en pacientes con hiperlipidemia y/o riesgo cardiovascular elevadoABC está contraindicado en pacientes con HLA-B*5701 positivo; cuando se prescriba se deben tomar las medidas necesarias para tratar de minimizar todos los FRCV modificablesMayor potencial de interacciones que otras pautas | |
| 3TC + LPV/r | Evitar en pacientes con hiperlipidemia y/o riesgo cardiovascular elevadoPuede ser una alternativa a la triple terapia convencional cuando no se pueda utilizar TDF ni ABCMayor potencial de interacciones que otras pautas | |
| RAL + DRV/r | No utilizar en pacientes con recuento de CD4 < 200 células/μlEvitar en los pacientes con CVP > 100.000 copias/mlPuede ser una alternativa a la triple terapia convencional cuando no se pueda utilizar TDF ni ABCMayor potencial de interacciones que otras pautas | |
| RAL + LPV/r | Evitar en pacientes con hiperlipidemia y/o riesgo cardiovascular elevadoNo utilizar en pacientes con recuento de CD4 < 200 células/μlEvitar en los pacientes con CVP > 100.000 copias/mlPuede ser una alternativa a la triple terapia convencional cuando no se pueda utilizar TDF ni ABCMayor potencial de interacciones que otras pautas | |
Cuando estén disponibles, se recomienda el uso de preparados que combinen fármacos a dosis fijas. No existe en la actualidad suficiente información que permita considerar como equivalentes terapéuticos a FTC y 3TC, por lo que el uso de uno u otro fármaco en los regímenes seleccionados depende fundamentalmente de la experiencia disponible en su uso conjunto con los otros fármacos de la combinación.
Los ensayos clínicos en los que se fundamenta la evidencia de cada pauta se referencian en el texto.
En los fármacos de una misma familia terapéutica e igual nivel de recomendación, el orden refleja la preferencia de los miembros del panel.
Los comentarios reflejan aspectos que se deben considerar en la elección de régimen, pero no pretenden ser una guía exhaustiva de las precauciones a tomar en el uso de los fármacos. Para mayor información se recomienda revisar el texto del documento, así como las fichas técnicas de los fármacos.
En otro apartado de estas guías se tratan aspectos de precio y de costes de los diferentes regímenes terapéuticos. Simultáneamente con las guías se publica un artículo en el que se hace un análisis formal de coste/eficacia de las pautas recomendadas.
El uso de cobicistat (COBI) como potenciador de DRV o ATV ha sido aprobado por la EMA, aunque en el momento de redactar estas guías COBI no está comercializado en España como fármaco independiente. También se ha autorizado una coformulación de DRV/COBI en un único comprimido, que tampoco está disponible en nuestro país.
Solo en el caso de que no puedan utilizarse tenofovir (TDF) o ABC, se pueden recomendar combinaciones de 2 FAR que excluyen uno o ambos ITIAN, pero en el momento actual ninguna puede ser considerada preferente para el inicio del TAR.
Recomendación- –
Pueden utilizarse las combinaciones de 2 ITIAN + 1 ITINN, 2 ITIAN + 1 IP/r o 2 ITIAN + 1 INI como TAR de inicio (los FAR preferentes se detallan más adelante, tabla 3) (A-I).
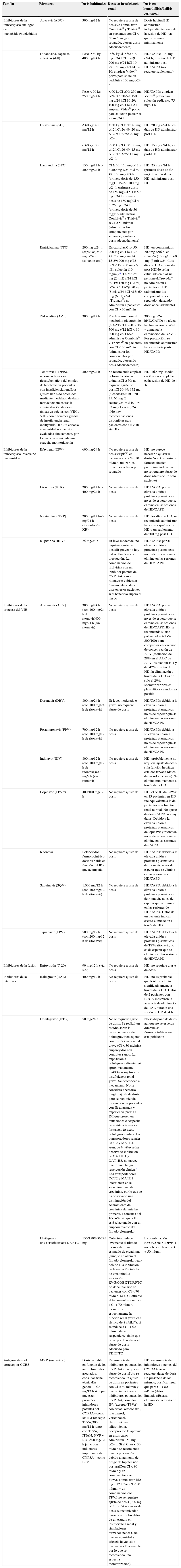
En España están comercializados 6 ITIAN: zidovudina (ZDV o AZT), didanosina (ddI), estavudina (d4T), lamivudina (3TC), emtricitabina (FTC) y ABC. También se dispone de un análogo de nucleótido (TDF). A efectos prácticos, la abreviatura ITIAN incluye también al TDF en esta guía.
Se consideran como combinaciones de ITIAN de elección las formadas por TDF/FTC y por ABC/3TC, que deberían administrase siempre que sea posible en preparados coformulados. No existe en la actualidad suficiente información que permita considerar como equivalentes terapéuticos a FTC y 3TC, por lo que el uso de uno u otro ITIAN en los regímenes seleccionados depende fundamentalmente de la experiencia disponible en su uso conjunto con los otros FAR de la combinación.
La mayor toxicidad relacionada con el uso de ZDV, ddI y d4T no permite recomendar su uso en ninguna pauta de inicio.
Combinaciones con TDF/FTC frente a combinaciones con ABC/3TCEn el ensayo clínico ACTG 520242 se comparó de forma ciega el inicio de TAR con ABC/3TC o TDF/FTC en 1.857 pacientes. Los participantes fueron aleatorizados además a recibir atazanavir/ritonavir (ATV/r) o efavirenz (EFV) de forma abierta. Entre los pacientes con CVP basal igual o mayor de 100.000 copias/ml, tanto el tiempo hasta el FV como el tiempo hasta el primer efecto adverso de grado 3-4 fueron significativamente más cortos en el brazo de ABC/3TC que en el brazo de TDF/FTC, lo que ocasionó la interrupción del estudio para los pacientes en este estrato de CVP. En los pacientes con CVP menor de 100.000 copias/ml no hubo diferencias en eficacia virológica entre ABC/3TC y TDF/FTC, independientemente de que se administraran con ATV/r o EFV43,44.
El estudio ASSERT45 es un ensayo clínico abierto en el que se compararon los perfiles de seguridad de TDF/FTC y ABC/3TC (ambos administrados con EFV) en pacientes con el HLA-B*5701-negativo. Aunque no fue diseñado para comparar eficacia, se observó que la proporción de pacientes con CVP inferior a 50 copias/ml a las 48 semanas fue significativamente superior con TDF/FTC que con ABC/3TC.
A diferencia de lo observado en el estudio ACTG 5202, en el estudio HEAT46 (doble ciego de 96 semanas de duración) no se observaron diferencias en la eficacia virológica ni en la respuesta inmunológica de ABC/3TC comparado con TDF/FTC cuando se combinan con lopinavir/ritonavir (LPV/r), con independencia de la CVP de partida.
Tres estudios en faseiii diseñados para comparar el TAR de inicio con dolutegravir (DTG), frente a otros FAR recomendados (EFV en el estudio SINGLE47, raltegravir [RAL] en el estudio SPRING248 o darunavir/ritonavir [DRV/r] en el estudio FLAMINGO49) han mostrado una eficacia similar de ABC/3TC o TDF/FTC. Sin embargo, dichos ensayos clínicos no permiten establecer comparaciones formales puesto que, o bien la elección de ITIAN no fue aleatorizada, quedando a criterio del investigador48,49, o las distintas combinaciones de ITIAN iban asociadas a un tercer fármaco también distinto47.
Finalmente, en un metaanálisis de ensayos clínicos no se observaron diferencias en la eficacia virológica de los tratamientos basados en ABC o TDF, aunque sí una mayor frecuencia de discontinuaciones debida a efectos adversos con ABC50.
En el caso de seleccionar un régimen basado en ABC, se debe realizar siempre previamente la determinación del HLAB*5701 del paciente, que de ser positivo contraindicaría su uso.
Recomendaciones- –
Las combinaciones de ITIAN de elección para regímenes de TAR de inicio son TDF/FTC o ABC/3TC (A-I). Se recomienda su uso en coformulaciones (A-II).
- –
La combinación TDF/FTC debe evitarse en pacientes con insuficiencia renal (A-II).
- –
La combinación ABC/3TC se debe evitar en pacientes con CVP elevada (más de 100.000 copias/ml) cuando se combina con un ITINN o un IP/r distinto de LPV/r (A-II).
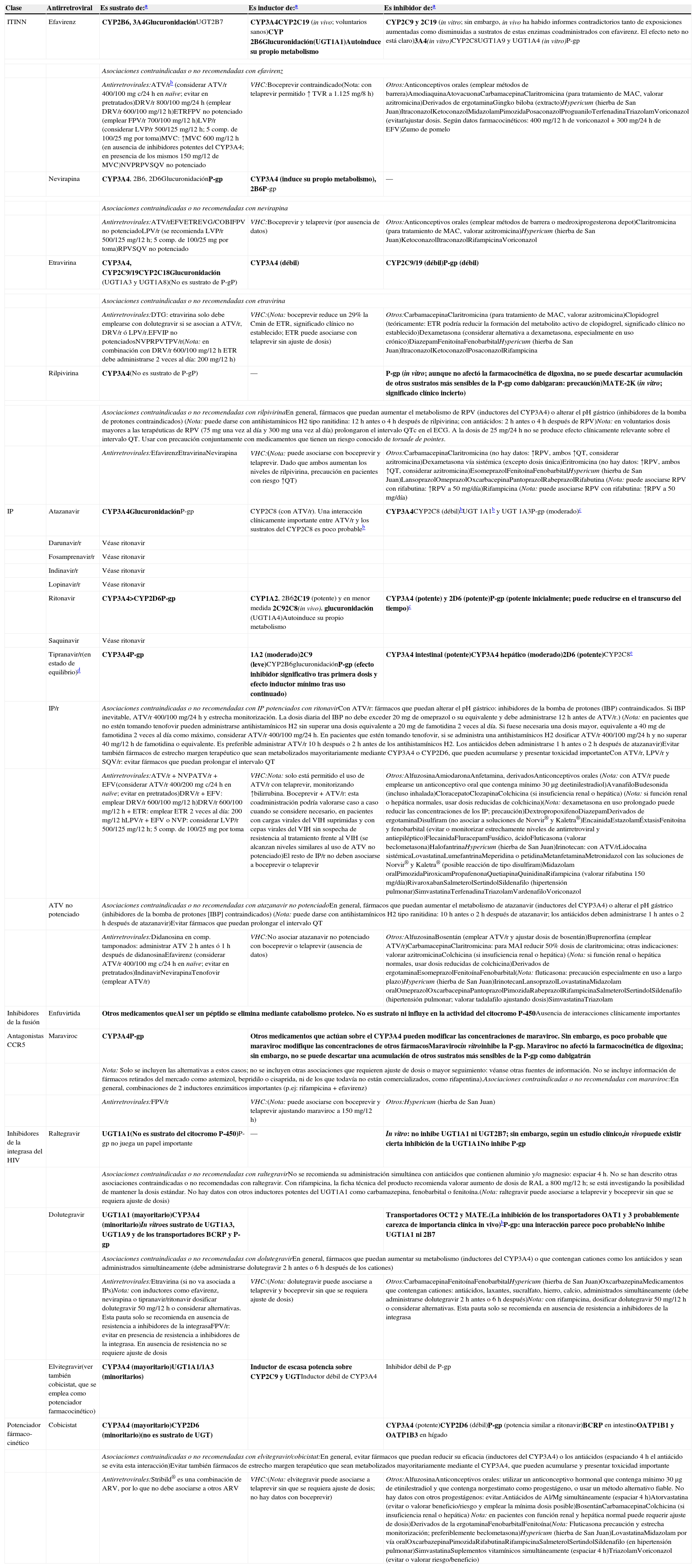
En España hay 4 ITINN comercializados: nevirapina (NVP), EFV, etravirina (ETR) y rilpivirina (RPV). Son inductores de algunas isoenzimas del citocromo P450, pudiendo interaccionar con otros fármacos. EFV se administra una vez al día (QD) (un comprimido de 600mg/día, existiendo una presentación coformulada con TDF/FTC en un solo comprimido). Su principal limitación es la frecuente aparición de síntomas relacionados con el sistema nervioso central (SNC), que aparecen al empezar su toma y que, aunque suelen ser leves y transitorios, pueden dar lugar a discontinuaciones del TAR. Esto hace que sea un FAR a evitar en algunas circunstancias, tales como trabajos de riesgo que requieran concentración, turnos laborales cambiantes o trastornos psiquiátricos no controlados. En un análisis de 4 grandes ensayos clínicos independientes se ha encontrado un mayor riesgo de intentos de suicidio y de suicidios consumados en los pacientes que recibían EFV (2,90 por 1.000 personas-año vs. 1,22 por 1.000 personas-año)51. Con EFV también existe riesgo de exantema durante las primeras semanas, lo que se debe advertir al paciente. EFV debe evitarse en mujeres gestantes o con deseo de gestación por el potencial riesgo de teratogenicidad.
RPV se administra QD (un comprimido de 25mg/día, existiendo también una presentación coformulada con TDF/FTC en un solo comprimido); debe administrase con una comida y está contraindicado el uso de inhibidores de la bomba de protones. NVP se puede administrar tanto QD (400mg/día, en comprimido de liberación retardada) o 2 veces al día (BID) (200mg/12h), aunque durante los primeros 14 días se administra un comprimido de 200mg al día. Está contraindicada en mujeres con más de 250 linfocitos CD4+/μl y en varones con más de 400 linfocitos CD4+/μl, por presentar mayor riesgo de RHS. ETR (un comprimido de 200mg/12h) no está aprobada por la EMA para el TAR de inicio.
EFV se ha comparado en ensayos clínicos con otros ITINN. Tres ensayos clínicos han comparado el uso de EFV frente a RPV, ambos combinados con 2 ITIAN52-54. Los estudios ECHO52 y THRIVE53 incluyeron a pacientes adultos sin TAR previo y sin MR en el estudio genotípico basal. Los participantes fueron aleatorizados a recibir de forma ciega RPV o EFV junto a 2 ITIAN (TDF/FTC coformulado en el estudio ECHO y una pareja de ITIAN seleccionada por los investigadores en el estudio THRIVE, que en el 60% de los casos fue también TDF/FTC). El análisis combinado de ambos estudios a las 96 semanas demostró la noinferioridad de RPV con respecto a EFV. La tasa de FV fue, sin embargo, superior con RPV en el subgrupo de pacientes con CVP al inicio del TAR mayor de 100.000 copias/ml (17,6 vs. 7,6%), por lo que no se recomienda el uso de RPV/TDF/FTC en estos pacientes. El FV con RPV se asoció con mayor frecuencia a resistencia genotípica a otros ITINN y a ITIAN (especialmente por selección de las mutaciones M184I y M184V). La tolerabilidad fue mejor con RPV, con un menor número de discontinuaciones por efectos adversos y menos efectos adversos relacionados con el SNC54.
En el tercer ensayo clínico (STaR)55 se compararon de forma abierta 2 pautas de TAR basadas en regímenes de un solo comprimido en pacientes sin TAR previo: RPV/TDF/FTC frente a EFV/TDF/FTC. Se demostró la noinferioridad de RPV/TDF/FTC frente a EFV/TDF/FTC en la población total tanto a las 48 como a las 96 semanas55. En el análisis de subgrupos RPV/TDF/FTC fue superior a EFV/TDF/FTC en los pacientes con CV igual o menor de 100.000 copias/ml, noinferior en los pacientes con CVP superior a 100.000 copias/ml e inferior en pacientes con CVP mayor de 500.000 copias/ml. Los pacientes tratados con RPV, en relación con los tratados con EFV, tuvieron una menor frecuencia de retirada del tratamiento por efectos adversos, una menor incidencia de efectos adversos del SNC y una menor frecuencia de efectos adversos psiquiátricos. No se dispone de comparaciones de RPV con otros FAR en el TAR de inicio.
NVP se ha comparado con EFV en el estudio 2NN56, en el que no logró demostrar la noinferioridad. Además, el uso de NVP se asoció con mayor toxicidad.
EFV ha demostrado una eficacia superior frente a LPV/r57, saquinavir/ritonavir (SQV/r)58 o amprenavir/ritonavir (APV/r)59. El único IP/r que hasta el momento ha mostrado una eficacia equiparable a EFV es ATV/r. En el estudio ACTG 520242 la eficacia virológica resultó similar en los tratados con ATV/r que en los tratados con EFV, independientemente que recibieran ABC/3TC o TDF/FTC. Entre los pacientes que experimentaron FV, la emergencia de cepas con MR fue significativamente menor en los pacientes tratados con ATV/r que entre los tratados con EFV. El tiempo hasta el primer evento de seguridad y el primer evento de tolerabilidad fue significativamente más largo para los pacientes con ATV/r que para los pacientes con EFV cuando la pareja de ITIAN era ABC/3TC, pero no hubo diferencias en seguridad ni en tolerabilidad entre ATV/r y EFV cuando la pareja de ITIAN era TDF/FTC.
Es en la comparación con los INI donde se puso de manifiesto que es posible superar la eficacia demostrada con EFV. El estudio STARTMRK60 comparó el uso de EFV frente a RAL, asociados con TDF/FTC. El estudio mantuvo el diseño ciego durante los 5 años de seguimiento. RAL resultó noinferior a EFV durante los 3 primeros años de tratamiento, pero se mostró superior a partir del cuarto año. Además, se observó una mayor rapidez en la supresión virológica con RAL, efecto que posteriormente se ha observado con todas las pautas que incluyen un INI, pero los datos de seguimiento no permiten afirmar que esta circunstancia tenga relevancia en la evolución clínica.
La combinación de EFV/FTC/TDF también ha sido evaluada en el estudio GS-US-236-010261, un ensayo clínico aleatorizado y doble ciego que comparó 2 regímenes administrados en un único comprimido: elvitegravir/cobicistat/FTC/TDF (EVG/COBI/FTC/TDF) y EFV/FTC/TDF. Se han comunicado datos a 3años que confirman la noinferioridad de la pauta con EVG/COBI/FTC/TDF frente a EFV/FTC/TDF. No hubo diferencias significativas en lo que respecta a retirada del TAR por efectos adversos entre ambos brazos de tratamiento. El incremento en la creatinina sérica en la semana 48 fue mayor en los tratados con EVG/COBI/FTC/TDF que en los tratados con EFV/FTC/TDF, lo que se ha relacionado con cambios en la secreción tubular de creatinina y no a un efecto directo sobre el FGe. Cabe señalar que en este estudio se excluyeron los pacientes con un FGe inferior a 70ml/min y también la baja proporción de pacientes en fase avanzada (12% con menos de 200 CD4+/μl), lo que limita la generalización de los resultados a estos subgrupos.
En el estudio SINGLE47,62 se compararon de forma ciega EFV/FTC/TDF y DTG+ABC/3TC. Las tasas de respuesta en la semana 48 (CVP inferior a 50 copias/ml por un análisis ITT, según el algoritmo snapshot de la FDA) fueron del 88% de los tratados con DTG+ABC/3TC y del 81% de los tratados con EFV/FTC/TDF (diferencia: 7,4%; IC95%: 2,5-12,3%), y en la semana 144 fueron del 71 y del 63%, respectivamente (diferencia: 8,3%; IC95%: 2,0-14,6%), confirmándose la superioridad de DTG+ABC/3TC sobre EFV/FTC/TDF. La proporción de FV fue similar en ambos brazos (9 y 8% al tercer año). La proporción de interrupciones del TAR por efectos adversos fue, sin embargo, mayor en el brazo de EFV/FTC/TDF (14%) que en el brazo de DTG+ABC/3TC (4%). La detección de mutantes resistentes tras el FV fue muy baja en ambos brazos de tratamiento, particularmente en el de DTG+ABC/3TC, en el que no se detectaron MR al INI en ningún caso. Al igual que el estudio anterior, la proporción de pacientes en fase avanzada fue baja (14%), por lo que tiene la misma limitación que aquél.
NVP se ha comparado con ATV/r, ambos combinados con TDF/FTC, en el ensayo ARTEN63, demostrándose la noinferioridad de NVP frente a ATV/r a las 48 semanas. La frecuencia de efectos adversos graves fue similar en ambos brazos, aunque las retiradas motivadas por efectos adversos resultaron más frecuentes con NVP que con ATV/r. Ninguno de los 28 pacientes con FV del brazo de ATV/r seleccionó cepas de VIH-1 con MR, mientras que ello ocurrió en 29 de los 44 pacientes con FV del brazo de NVP.
Recomendaciones- –
La combinación de EFV/TDF/FTC se considera una opción alternativa de tratamiento (A-I).
- –
La combinación de EFV+ABC/3TC debe evitarse en pacientes con CVP mayor de 100.000 copias/ml (B-I).
- –
EFV está contraindicado durante el primer trimestre de la gestación. Se recomienda considerar otras opciones en mujeres que no utilicen métodos anticonceptivos eficaces. Se debe evitar en pacientes con alteraciones neuropsiquiátricas no estabilizadas y en pacientes que realicen tareas peligrosas si presentan síntomas de somnolencia, mareos o trastornos de la concentración (A-III).
- –
NVP está contraindicado en mujeres con cifras de linfocitos CD4+ superiores a 250 células/μl y en varones con cifras superiores a 400 células/μl (A-II).
- –
RPV no debe utilizarse en pacientes con CVP mayor de 100.000 copias/ml (A-II).
- –
En pacientes con CVP inferior a 100.000 la combinación RPV/TDF/FTC se considera un régimen de elección (A-I).
En el TAR de inicio solo se pueden usar IP cuando van potenciados con dosis bajas de ritonavir o COBI. En la actualidad los IP potenciados disponibles en la clínica son 6: ATV, DRV, LPV, fosamprenavir (FPV), SQV y tipranavir (TPV), aunque este último está aprobado por la EMA solamente para pacientes pretratados. Los IP son inductores e inhibidores del citocromo P450 y frecuentemente pueden originar interacciones farmacológicas. La elección final del IP se basará en datos de eficacia, tolerabilidad, posología y farmacocinética. LPV se administra en comprimidos coformulados con ritonavir. COBI se ha autorizado por la EMA para potenciar ATV o DRV, habiéndose aprobado recientemente la utilización de DRV/COBI en un único comprimido coformulado.
Los IP se caracterizan por una elevada barrera genética frente a la resistencia, que dificulta la selección de MR aun en situaciones desfavorables, como una baja adherencia. Por otra parte, son los FAR con peor perfil metabólico, asociándose a hiperlipidemia, aunque en menor medida en el caso de ATV y DRV.
LPV fue el primer IP desarrollado para utilizar potenciado y ha sido el referente de esta familia durante años. El resto de IP se han comparado con LPV/r en diversos ensayos clínicos demostrando la noinferioridad de FPV/r64, SQV/r65, ATV/r66 y DRV/r67. SQV/r y FPV/r tienen en la actualidad un uso muy limitado debido a que no aportan ventajas significativas en cuanto a simplicidad o tolerabilidad con los otros FAR de la familia.
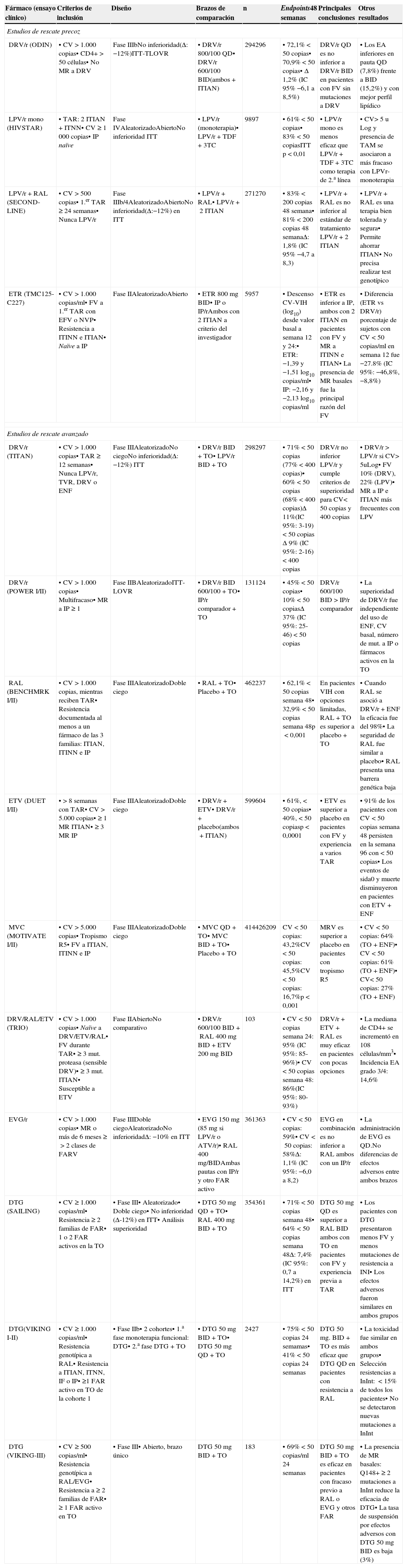
DRV/r se utiliza en el TAR de inicio en pautas QD (un comprimido de 800mg/día potenciado con 100mg de ritonavir o 150mg de COBI). El estudio ARTEMIS67 comparó DRV/r (800/100mg, QD) frente a LPV/r en 689 pacientes que recibieron además TDF/FTC coformulados. A las 48 semanas DRV/r resultó noinferior a LPV/r. Los pacientes tratados con DRV/r presentaron menos diarrea de grado 2-4 y menores elevaciones de colesterol y triglicéridos que los tratados con LPV/r. A las 96 semanas, DRV/r resultó superior a LPV/r (en el análisis TLOVR, no en el snapshot). Un 4% de los pacientes de la rama de DRV/r y un 9% de los la rama de LPV/r abandonaron el tratamiento asignado. DRV/r se ha comparado también con ATV/r y con RAL (los 3 administrados junto a TDF/FTC) en el estudio ACTG525768, que incluyó 1.809 pacientes. Tras 96 semanas, los 3 regímenes fueron equivalentes en eficacia virológica (DRV/r 85%, ATV/r 87% y RAL 90%), pero tanto en el análisis conjunto de la respuesta virológica y la tolerabilidad, como en el análisis snapshot, DRV/r fue inferior a RAL y superior a ATV/r (DRV/r 73%, ATV/r 63%, RAL 89%). DRV/r se ha comparado con DTG en un ensayo clínico de TAR de inicio (FLAMINGO)49, en el que se observó menor eficacia a 48 semanas del DRV/r, hecho motivado fundamentalmente por una mayor tasa de discontinuaciones por causas no relacionadas con el fármaco y efectos adversos.
ATV/r se ha comparado también con LPV/r y con EVG/COBI en el TAR de inicio. El estudio CASTLE66 comparó la combinación TDF/FTC+ATV/r QD con la combinación TDF/FTC+LPV/r BID, demostrando la noinferioridad de ATV/r frente a LPV/r a 48 y a 96 semanas. ATV/r mostró mejor perfil lipídico (colesterol total, triglicéridos y colesterol no-HDL). La ictericia e hiperbilirrubinemia fueron más frecuentes en el grupo de ATV/r, mientras que la diarrea y las náuseas lo fueron en el grupo de LPV/r. No hubo diferencias significativas en abandonos entre los 2 brazos de tratamiento. El estudio GS-US-236-010369, un ensayo clínico aleatorizado a doble ciego, comparó EVG/COBI/FTC/TDF (coformulados en un solo comprimido) y ATV/r+FTC/TDF, confirmando la noinferioridad de EVG/COBI/FTC/TDF frente a ATV/r+FTC/TDF. En el grupo de EVG/COBI/FTC/TDF se efectuó genotipificación del VIH-1 tras el FV en 12 pacientes, en 5 de los cuales se objetivaron mutaciones, que conferían resistencia a los INI en 4 de ellos (Q148R en 2, N155H en 2, T66I en uno y E92Q en uno). En el grupo de ATV/r+FTC/TDF se realizó un estudio genotípico del VIH-1 en 8 pacientes con FV, en ninguno de los cuales se detectaron MR. Ambos regímenes se toleraron bien, y las interrupciones del tratamiento por efectos adversos fueron escasas en ambos brazos.
La utilización de COBI como potenciador de los IP se valoró en el estudio 11470, en el que se comparó de forma ciega COBI frente a ritonavir como potenciador de ATV+TDF/FTC, en 692 pacientes sin TAR previo y con un FGe igual o superior a 70ml/min. Se demostró la noinferioridad del brazo potenciado con COBI, aunque el estudio no fue capaz de demostrar ventajas significativas de tolerabilidad con el nuevo potenciador. COBI ha sido aprobado por la EMA como potenciador de ATV o DRV a dosis de 150mg/día.
En regímenes dobles con un IP/r junto a otro FAR en el TAR de inicio (biterapia), los mejores resultados hasta este momento se han publicado con la combinación LPV/r+3TC, que ha demostrado la noinferioridad frente a LPV/r+2 ITIAN71, independientemente de la CVP, por lo que podría considerarse una alternativa a la triple terapia convencional cuando no se pueda utilizar TDF ni ABC. La combinación LPV/r+RAL ha demostrado la noinferioridad sobre LPV/r+TDF/FTC72, pero los pacientes incluidos en el estudio tenían CVP muy bajas y no es posible generalizar sus resultados. DRV/r+RAL también ha demostrado la noinferioridad con respecto a la triple terapia (DRV/r+TDF/FTC)73 en un estudio independiente con 805 pacientes, pero en el subgrupo de pacientes con cifras de linfocitos CD4+ inferiores a 200 células/μl la terapia doble presentó menor eficacia que la triple terapia convencional.
Recomendaciones- –
Los regímenes basados en IP recomendados son: DRV/r (o DRV/COBI) QD+TDF/FTC y ATV/r (o ATV/COBI) QD+TDF/FTC (A-I). La combinación de ATV/r (o ATV/COBI)+ABC/3TC también se recomienda, pero se debe evitar en pacientes con CVP superior a 100.000 copias/ml (A-I).
- –
Otras pautas con IP incluyen LPV/r, BID o QD, +TDF/FTC o ABC/3TC (B-I). Es posible utilizar también la combinación DRV/r (o DRV/COBI)+ABC/3TC, aunque no ha sido formalmente investigada en ningún ensayo clínico (B-III).
- –
ATV y DRV pueden ser potenciados indistintamente con 100mg de ritonavir o 150mg de COBI (B-II).
- –
LPV/r+3TC, LPV/r+RAL y DRV/r+RAL pueden ser una alternativa a la triple terapia convencional cuando no se pueda utilizar TDF ni ABC (B-I). Las pautas dobles sin ITIAN (DRV/r o LPV/r+RAL) no deben utilizarse como tratamiento de inicio en pacientes en fase avanzada (cifra de linfocitos CD4+ inferior a 200 células/μl y/o CVP superior a 100.000 copias/ml) (A-I).
Los INI aprobados por la EMA para el TAR de inicio son: RAL, EVG y DTG. RAL fue el primer INI comercializado y con el que se tiene una mayor experiencia. Se utiliza en una pauta administrada 2 veces al día (400mg BID), puesto que la administración de los 2 comprimidos juntos una única vez al día demostró una menor eficacia en un ensayo clínico de pacientes sin TAR previo74. Combinado con TDF/FTC, RAL BID ha demostrado ser superior a DRV/r68 y a ATV/r68, y noinferior a EFV, alcanzando eficacia superior al cuarto y quinto año de seguimiento60. EVG requiere potenciación farmacológica. Se ha comercializado coformulado con COBI (que hace la función de potenciador) +TDF/FTC en un solo comprimido. EVG/COBI/TDF/FTC ha demostrado la noinferioridad con respecto a EFV/TDF/FTC61 y a ATV/r+TDF/FTC69 (véanse los apartados de ITINN e IP) en estudios con una duración de 3años. Esta combinación está contraindicada en pacientes con un FGe inferior a 70ml/min y debe utilizarse con precaución en aquellos con un FGe entre 70 y 90ml/min. DTG se administra como un comprimido de 50mg una vez al día (en algunas circunstancias se debe administrar 2 veces al día) y no necesita potenciación. Se ha comparado en estudios en faseiii con FAR de las 3 familias que en algún momento han sido recomendados en el TAR de inicio, mostrando una eficacia superior a EFV47,62 y a DRV/r49 (véanse los apartados de ITINN e IP). La comparación con RAL se realizó en un estudio doble ciego (SPRING-2), que confirmó la noinferioridad de DTG. Cabe destacar que no se ha seleccionado ninguna MR a DTG en ninguno de los 3 ensayos clínicos en faseiii de inicio de TAR, lo que hace pensar que DTG presenta una barrera genética frente a la resistencia superior a la de otros INI, aunque se deberá confirmar cuando se disponga de mayor experiencia y seguimiento.
Recomendaciones- –
DTG combinado con TDF/FTC o ABC/3TC y RAL combinado con TDF/FTC se consideran pautas preferentes como TAR de inicio (A-I).
- –
La combinación EVG/COBI/TDF/FTC pueden utilizarse como TAR de inicio, pero no debe usarse en pacientes con FGe menor de 70ml/min) (A-I).
- –
La combinación de RAL con ABC/3TC se considera una alternativa en el tratamiento de inicio, pues dispone de menor evidencia (A-I).
En este capítulo se revisan las opciones de cambio de regímenes de TAR en pacientes que tienen la CVP suprimida.
Motivos para cambiar un tratamiento antirretroviral eficazExisten muchos motivos para cambiar un TAR eficaz: toxicidad, nuevas comorbilidades, interacciones farmacológicas, disminución del número de pastillas o dosis diarias, requerimientos dietéticos, embarazo y coste del propio TAR. El cambio puede ser proactivo, cuando se realiza preventivamente, o reactivo, cuando el régimen actual ha dejado de ser el ideal para el paciente debido a alguno de los motivos reseñados.
Definición de carga viral suprimidaConsideramos que la CVP está suprimida cuando es menor de 50 copias/ml y está confirmada. La mayoría de los ensayos clínicos de cambio de TAR han incluido pacientes que habían mantenido este nivel de supresión virológica durante al menos 6 meses, por lo que sus resultados son aplicables preferiblemente a pacientes con un tiempo de supresión similar. Como norma general, cuanto más prolongado sea el período de supresión virológica, menos probable es que el cambio de TAR se asocie a FV.
Objetivo del cambio del tratamiento antirretroviral eficazEl objetivo es mantener la supresión virológica y optimizar el TAR de acuerdo a las características y a los deseos de cada paciente individual.
Circunstancias que obligan a cambiar el tratamiento antirretroviral eficazEl cambio proactivo es obligado cuando evidencias sólidas avalan que el paciente tiene más riesgo de presentar un efecto adverso grave o irrecuperable si se mantiene el TAR actual que si se cambia. Un ejemplo paradigmático es la lipoatrofia causada por análogos timidínicos. El cambio reactivo es obligado si el efecto adverso va a desaparecer tras el cambio de TAR, como, por ejemplo, los efectos adversos del SNC causados por EFV.
Los efectos adversos que ocasionan el cambio precoz del TAR inicial en un paciente naïve son de tal intensidad que el cambio se realiza frecuentemente antes de haberse alcanzado la supresión de la replicación viral. Es obvio que si un paciente tiene la CVP indetectable es porque es capaz de continuar tomando un TAR eficaz. El clínico no debe olvidar que en ocasiones ese nivel de adherencia se consigue gracias al sacrificio del paciente, que es capaz de sobrellevar efectos adversos que pueden ser erróneamente entendidos como inevitables. El médico no debe asumir que un TAR es óptimo para su paciente solo porque la CVP está suprimida. Este comité recomienda que en todas las revisiones el clínico pregunte con detalle sobre el esfuerzo que necesita el paciente para adherirse al TAR pautado.
Cambio de antirretroviralesConsideraciones virológicasTras el cambio del TAR (independientemente de las clases de FAR implicadas), el mantenimiento de la supresión virológica es la norma en los pacientes sin historia de FV. Cambiar el TAR es más complicado en pacientes con FV previos que pueden haber causado MR ya archivadas. En esta situación el clínico debe diseñar una nueva pauta cuya barrera genética frente a la resistencia no sea inferior a la previa. Esta precaución es crítica cuando se cambia un TAR que incluye IP/r. En esta situación el clínico debe diseñar una pauta que tenga en cuenta una eventual resistencia archivada, bien sea confirmada o bien sea sospechada.
Recomendación- –
El cambio desde un pauta con 2 ITIAN más un IP/r a 2 ITIAN más un ITINN, RAL, EVG/COBI o ATV no potenciado con ritonavir solo debe hacerse si se puede garantizar la actividad antiviral de los 2 ITIAN y la del tercer fármaco acompañante (A-I). Sin olvidar que el objetivo prioritario es mantener la supresión virológica, el clínico debe realizar una evaluación minuciosa del perfil de toxicidades, interacciones farmacológicas, restricciones dietéticas y actividad sobre el VHB (si fuera necesario) del nuevo régimen.
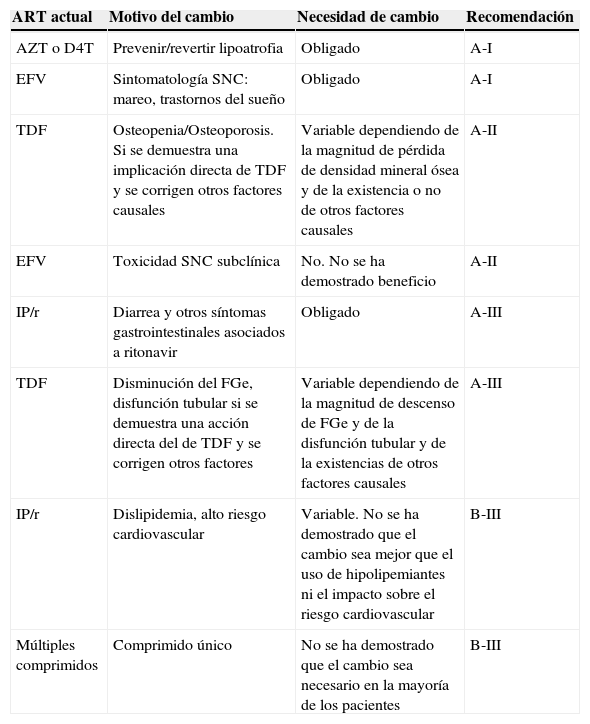
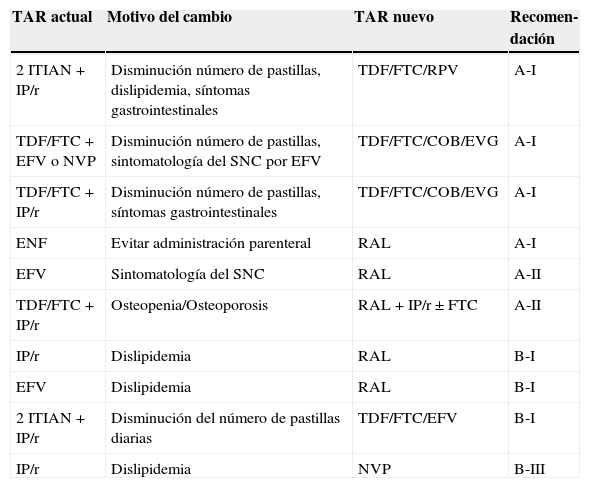
Se considera que existen datos suficientes para hacer una recomendación fuerte de cambio de antirretrovirales (tabla 4) para:
- –
Cambio proactivo de análogos timidínicos por ABC o TDF. En esta situación hay evidencia de alta calidad que avala que los beneficios del cambio superan ampliamente al riesgo de aparición o empeoramiento de lipoatrofia, si se continúa el tratamiento con análogos timidínicos.
- –
Cambio reactivo de EFV en pacientes con efectos adversos del SNC.
- –
Cambio reactivo de IP/r si existe diarrea que interfiere con la calidad de vida del paciente
- –
Cambio reactivo de TDF en pacientes con disminución del FGe o disfunción tubular no explicable por otras causas. No hay datos para precisar un umbral de FGe o parámetros de tubulopatía a partir del cual el cambio sea obligado. Recomendamos emplear el juicio clínico teniendo en cuenta función renal/tubular antes del inicio de TDF, edad y existencia de otros factores que puedan empeorar la función renal.
- –
Cambio reactivo de TDF en pacientes con osteoporosis u osteomalacia no explicable por otros motivos. No hay datos para precisar un umbral de densidad mineral ósea (DMO) a partir del cual el cambio sea obligado. Es preciso descartar múltiples causas secundarias, por ejemplo hipovitaminosis D, antes de atribuir la disminución de la DMO exclusivamente a TDF.
Recomendaciones sobre la necesidad de cambio del tratamiento antirretroviral asumiendo que mantendrá la supresión virológica
| ART actual | Motivo del cambio | Necesidad de cambio | Recomendación |
|---|---|---|---|
| AZT o D4T | Prevenir/revertir lipoatrofia | Obligado | A-I |
| EFV | Sintomatología SNC: mareo, trastornos del sueño | Obligado | A-I |
| TDF | Osteopenia/Osteoporosis. Si se demuestra una implicación directa de TDF y se corrigen otros factores causales | Variable dependiendo de la magnitud de pérdida de densidad mineral ósea y de la existencia o no de otros factores causales | A-II |
| EFV | Toxicidad SNC subclínica | No. No se ha demostrado beneficio | A-II |
| IP/r | Diarrea y otros síntomas gastrointestinales asociados a ritonavir | Obligado | A-III |
| TDF | Disminución del FGe, disfunción tubular si se demuestra una acción directa del de TDF y se corrigen otros factores | Variable dependiendo de la magnitud de descenso de FGe y de la disfunción tubular y de la existencias de otros factores causales | A-III |
| IP/r | Dislipidemia, alto riesgo cardiovascular | Variable. No se ha demostrado que el cambio sea mejor que el uso de hipolipemiantes ni el impacto sobre el riesgo cardiovascular | B-III |
| Múltiples comprimidos | Comprimido único | No se ha demostrado que el cambio sea necesario en la mayoría de los pacientes | B-III |
Este panel distingue entre la fuerza de la recomendación para cambiar el TAR o para priorizar una pauta alternativa. Recomendamos que el clínico consulte la tabla 4 para intentar responder a la pregunta ¿debo cambiar el TAR?, y una vez establecida la necesidad de cambio, las tablas 5 y 6 gradúan la evidencia para recomendar una nueva pauta.
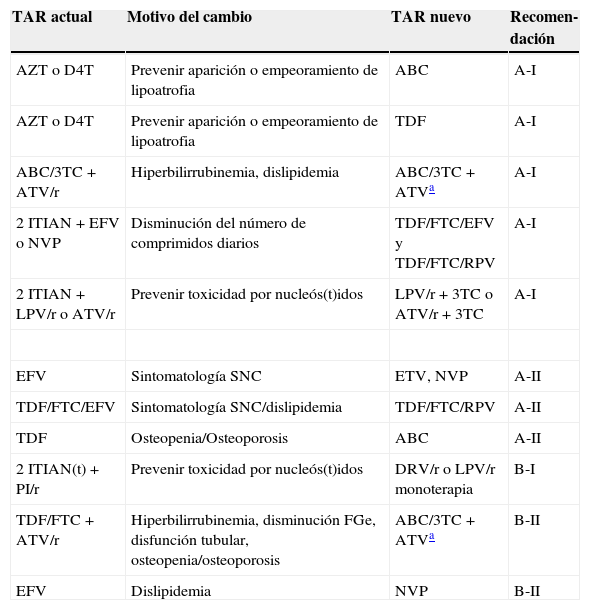
Recomendaciones sobre cambios entre fármacos antirretrovirales de la misma clase, según el motivo de cambio y ordenadas por la evidencia sobre la eficacia del cambio
| TAR actual | Motivo del cambio | TAR nuevo | Recomen-dación |
|---|---|---|---|
| AZT o D4T | Prevenir aparición o empeoramiento de lipoatrofia | ABC | A-I |
| AZT o D4T | Prevenir aparición o empeoramiento de lipoatrofia | TDF | A-I |
| ABC/3TC + ATV/r | Hiperbilirrubinemia, dislipidemia | ABC/3TC + ATVa | A-I |
| 2 ITIAN + EFV o NVP | Disminución del número de comprimidos diarios | TDF/FTC/EFV y TDF/FTC/RPV | A-I |
| 2 ITIAN + LPV/r o ATV/r | Prevenir toxicidad por nucleós(t)idos | LPV/r + 3TC o ATV/r + 3TC | A-I |
| EFV | Sintomatología SNC | ETV, NVP | A-II |
| TDF/FTC/EFV | Sintomatología SNC/dislipidemia | TDF/FTC/RPV | A-II |
| TDF | Osteopenia/Osteoporosis | ABC | A-II |
| 2 ITIAN(t) + PI/r | Prevenir toxicidad por nucleós(t)idos | DRV/r o LPV/r monoterapia | B-I |
| TDF/FTC + ATV/r | Hiperbilirrubinemia, disminución FGe, disfunción tubular, osteopenia/osteoporosis | ABC/3TC + ATVa | B-II |
| EFV | Dislipidemia | NVP | B-II |
Recomendaciones sobre cambios a fármacos antirretrovirales de una nueva clase, según el motivo de cambio y ordenadas por la evidencia sobre la eficacia del cambio
| TAR actual | Motivo del cambio | TAR nuevo | Recomen-dación |
|---|---|---|---|
| 2 ITIAN + IP/r | Disminución número de pastillas, dislipidemia, síntomas gastrointestinales | TDF/FTC/RPV | A-I |
| TDF/FTC + EFV o NVP | Disminución número de pastillas, sintomatología del SNC por EFV | TDF/FTC/COB/EVG | A-I |
| TDF/FTC + IP/r | Disminución número de pastillas, síntomas gastrointestinales | TDF/FTC/COB/EVG | A-I |
| ENF | Evitar administración parenteral | RAL | A-I |
| EFV | Sintomatología del SNC | RAL | A-II |
| TDF/FTC + IP/r | Osteopenia/Osteoporosis | RAL + IP/r ± FTC | A-II |
| IP/r | Dislipidemia | RAL | B-I |
| EFV | Dislipidemia | RAL | B-I |
| 2 ITIAN + IP/r | Disminución del número de pastillas diarias | TDF/FTC/EFV | B-I |
| IP/r | Dislipidemia | NVP | B-III |
Solo se han incluido cambios para los que al menos existe un ensayo clínico pertinente y existe un análisis sobre el efecto adverso particular.
La recomendación es fuerte si existe al menos un ensayo clínico de calidad avalando una mejoría de balance riesgo/beneficio tras el cambio.
El cambio desde un pauta con 2 ITIAN más un IP/r a 2 ITIAN más un ITINN, RAL o EVG/COBI solo debe hacerse si se puede garantizar la actividad antiviral de los 2 ITIAN y la del tercer fármaco acompañante.
La recomendación para avalar el cambio de TAR puede ser débil, pero una vez que el cambio se ha decidido, la recomendación sobre la pauta a la que cambiar puede ser fuerte. Un ejemplo apropiado es el cambio desde múltiples pastillas a una pastilla única simplemente con el objetivo de simplificación. Aunque hay evidencia preliminar de que el TAR con pastilla única podría aumentar la adherencia y disminuir las hospitalizaciones75, en este momento el comité no considera que exista evidencia definitiva para hacer una recomendación fuerte que respalde este cambio en la mayoría de los pacientes. Sin embargo, si el clínico ha decidido que en un paciente la simplificación posológica está indicada, entonces el comité hace una recomendación fuerte sobre las pautas más apropiadas.
Cambio entre antirretrovirales de la misma clase (tabla 5). Inhibidores de la transcriptasa inversa análogos de nucleósidoCambio de d4T o AZT por TDF o ABCFundamento. Varios ensayos clínicos aleatorizados76-79 han demostrado que el cambio de d4T o AZT por ABC o TDF es seguro virológicamente y produce una mejoría subclínica o estabilización de la pérdida de grasa subcutánea. En pacientes cuyo HLA B*5701 sea negativo no parecen existir diferencias entre ABC y TDF en cuanto al mantenimiento de la supresión virológica.
Recomendación- –
El cambio proactivo de d4T o AZT a TDF o ABC es adecuado para prevenir o tratar de revertir la lipoatrofia asociada al tratamiento con análogos timidínicos (A-I).
Fundamento. Tres ensayos clínicos aleatorizados80-82 han demostrado que el cambio de ABC/3TC a TDF/FTC es seguro virológicamente y produce disminuciones de colesterol total (CT), colesterol LDL (cLDL) y triglicéridos (TG) sin cambios en el cociente CT/colesterol HDL (cHDL). La caída del FGe es mayor en los pacientes que cambian a TDF/FTC, especialmente si el tercer fármaco es un IP/r.
Recomendación- –
La asociación de ABC con un incremento en la incidencia de eventos cardiovasculares es muy controvertida. Este comité no puede hacer una recomendación en el momento actual sobre fuerza de la evidencia para realizar un cambio de ABC/3TC a TDF/FTC.
Fundamento. TDF se ha asociado con una disminución de la DMO independiente de la presencia o no de otros factores de riesgo de osteoporosis, que es mayor que con ABC83,84. En un pequeño ensayo clínico aleatorizado en pacientes con osteopenia u osteoporosis, el cambio de TDF a ABC se siguió de un incremento de la DMO en el fémur pero no en la columna vertebral85.
Recomendación- –
El cambio de TDF a ABC es una opción en pacientes con osteopenia u osteoporosis asociada al uso de TDF (A-II).
Fundamento. Dos ensayos clínicos no aleatorizados86,87 sugieren que el cambio es virológicamente seguro y puede mejorar la toxicidad persistente del SNC causada por el EFV así como los niveles de lípidos plasmáticos. La larga vida media de EFV y su capacidad de inducción enzimática sobre el CYP3A4 origina una disminución de las concentraciones plasmáticas de RPV que parece no tener trascendencia en pacientes que ya han alcanzado la supresión virológica. No hay datos de que este cambio se pueda realizar en pacientes con replicación de VIH-1 activa.
Recomendación- –
En pacientes con efectos adversos del SNC causados por TDF/FTC/EFV el cambio a TDF/FTC/RPV es una de las opciones que puede mejorar los síntomas neuropsicológicos asociados a EFV (A-II). No hay datos para recomendar un cambio proactivo en pacientes sin síntomas del SNC. No hay datos comparativos de este cambio frente al cambio por otros FAR que tampoco causan síntomas del SNC.
Fundamento. El cambio es seguro virológicamente y puede mejorar la toxicidad persistente del SNC causada por EFV88. En pacientes estables sin efectos adversos del SNC causados por EFV no hubo cambios apreciables en sintomatología del SNC89.
Recomendación- –
En pacientes con efectos adversos del SNC causados por EFV, el cambio a ETR es una opción que puede mejorar los síntomas neuropsicológicos asociados a EFV (A-II). No hay datos para recomendar un cambio proactivo en pacientes sin síntomas del SNC. No hay datos comparativos de este cambio frente al cambio por otros FAR que tampoco causan síntomas del SNC.
Fundamento. Un pequeño ensayo clínico aleatorizado y un subanálisis post hoc de un ensayo clínico sugieren que este cambio es virológicamente seguro y puede mejorar la toxicidad persistente del SNC y la elevación del cLDL causada por EFV. EFV y NVP son inductores del CYP3A4. Debido a su larga vida media y a su efecto inductor enzimático EFV, aunque se discontinúe, podría inducir niveles infraterapéuticos de NVP, si esta se inicia a dosis de 200mg una vez al día. Cuando se realiza el cambio a NVP puede utilizarse a dosis completa (400mg/día) sin necesidad de escalada de dosis durante 2 semanas90-92.
En pacientes con CVP suprimida no se aplica la limitación de uso de NVP según niveles de linfocitos CD4+ que se emplea en pacientes naïve. Múltiples estudios han demostrado que el riesgo de hepatotoxicidad de NVP no parece estar incrementado en pacientes con replicación viral suprimida, independientemente de la cifra de células CD4+93-95.
Recomendación- –
En pacientes con efectos adversos del SNC causados por EFV el cambio a NVP es una opción que puede mejorar los síntomas neuropsicológicos (A-II). No hay datos para recomendar un cambio proactivo en pacientes sin síntomas del SNC. No hay datos comparativos de este cambio frente al cambio por otros FAR que tampoco causan síntomas del SNC. El cambio también es una opción en pacientes con elevación del cLDL causada por EFV (A-II).
Fundamento. Un ensayo clínico abierto ha demostrado que en pacientes tratados con 2 ITIAN y EFV o NVP el cambio a TDF/FTC/EFV es seguro virológicamente96.
Recomendación- –
El cambio a TDF/FTC/EFV es una opción en pacientes que reciben TAR con EFV y NVP y desean disminuir el número de comprimidos diarios (A-II).
Fundamento. Este cambio solo se puede realizar si ABC/3TC es completamente activo. Un ensayo clínico ha demostrado que el cambio es virológicamente seguro y se asocia a una disminución de los niveles de bilirrubina, CT, cLDL y TG97.
Recomendación- –
En pacientes que reciben tratamiento con ABC/3TC+ATV/r, el cambio a ABC/3TC+ATV no potenciado es una opción de simplificación para pacientes en quienes no sea deseable continuar con ritonavir debido a que potencia la toxicidad de ATV (hiperbilirrubinemia), toxicidad (dislipidemia, diarrea) o riesgo de interacciones (A-I).
Fundamento. Un ensayo clínico aleatorizado ha demostrado que el cambio de TDF/FTC+ATV/r a ABC/3TC+ATV no potenciado es virológicamente seguro y se asocia a disminución de bilirrubina plasmática y mejoría de los biomarcadores de disfunción tubular renal y óseos. Se desconoce la relevancia clínica de los cambios de estos biomarcadores98.
Recomendación- –
En pacientes que reciben tratamiento con TDF/FTC+ATV/r el cambio a ABC/3TC+ATV no potenciado es una opción para pacientes en quienes se quiera evitar el uso tanto de TDF como de ritonavir (A-II).
Fundamento. Un pequeño ensayo clínico de brazo único en pacientes tratados con un IP/r más TDF/FTC y que presentaban disminución de la DMO (T-score de al menos −1) ha demostrado que el cambio de TDF a RAL (con o sin FTC) es seguro virológicamente y se asocia a una mejoría significativa de la DMO en fémur y cadera y de los marcadores de remodelación ósea99.
Recomendación- –
El cambio de TDF a RAL (en pacientes que además reciben un IP/r) es una opción en pacientes con disminución de la DMO (A-II).
Fundamento. En pacientes que toleran EFV, un ensayo clínico aleatorizado y doble ciego ha demostrado que el cambio de EFV a RAL mejora los niveles de lípidos (CT, cLDL y TG) y en algunos pacientes mejora las escalas de ansiedad y estrés, manteniendo la supresión virológica100.
Recomendación- –
El cambio de EFV a RAL es una opción en pacientes con efectos adversos del SNC causados por EFV (A-II). No hay datos para recomendar un cambio proactivo en pacientes sin síntomas del SNC. No hay datos comparativos de este cambio frente al cambio por otros FAR que tampoco causan síntomas del SNC.
- –
El cambio de EFV a RAL es una opción en pacientes con dislipidemia causada por EFV (A-I).
Fundamento. Un ensayo clínico aleatorizado y abierto ha demostrado que el cambio de TDF/FTC más un ITINN (en su amplia mayoría pacientes en su primer régimen de TAR, fundamentalmente basado en EFV) a TDF/FTC/COBI/EVG es seguro virológicamente. Los pacientes que discontinuaron EFV en este ensayo mejoraron significativamente los resultados en las escalas de sueños anormales, insomnio, mareo y ansiedad. Sin embargo, algunos efectos adversos como fatiga, tos, cefalea y náuseas aparecieron de manera más frecuente en el brazo de TDF/FTC/COBI/EVG101.
Recomendación- –
El cambio de TDF/FTC+EFV o NVP a TDF/FTC/COB/EVG coformulados es seguro virológicamente. Este cambio es una opción para los pacientes que quieran simplificar su régimen actual y puede mejorar los síntomas del SNC causados por EFV. No hay datos para recomendar un cambio proactivo en pacientes sin síntomas del SNC. No hay datos comparativos de este cambio frente al cambio por otros FAR que tampoco causan síntomas del SNC (A-I).
Fundamento. Un ensayo clínico aleatorizado ha demostrado que el cambio de enfuvirtida (ENF) a RAL es seguro virológicamente y los pacientes pueden evitar la administración parenteral102.
Recomendación- –
El cambio de ENF a RAL es un opción segura que evita las complicaciones de la administración parenteral de ENF (A-I).
Fundamento. Un ensayo clínico abierto ha demostrado que en pacientes tratados con 2 ITIAN y un IP/r el cambio a TDF/FTC/EFV es seguro virológicamente y se asocia con una mejoría de los niveles plasmáticos de TG y cHDL, pero con más efectos adversos del SNC96.
Recomendación- –
El cambio a TDF/FTC/EFV es una opción en pacientes que reciben TAR con IP/r y desean disminuir el número de pastillas diarias, pero es posible que experimenten efectos adversos del EFV sobre el SNC (B-I).
Fundamento. Múltiples estudios han demostrado que el cambio de IP (mayoritariamente no potenciados con ritonavir) a NVP es seguro virológicamente, aunque se asocia a una mayor incidencia de efectos adversos hepáticos. No hay datos relevantes para el cambio desde un IP/r103-105.
Recomendación- –
El cambio de un IP a NVP podría ser una opción en pacientes que reciben un IP/r cuando se quieran evitar los efectos adversos de ritonavir (B-III).
Fundamento. Un ensayo clínico abierto ha demostrado que el cambio desde 2 ITIAN más un IP/r a TDF/FTC/RPV coformulados es seguro virológicamente y se asocia a una mejoría en los niveles de CT, cLDL, cociente CT/cHDL y TG, y a una mejoría en los efectos adversos gastrointestinales causados por los IP/r106.
Recomendación- –
El cambio de 2 ITIAN más un IP/r a TDF/FTC/RPV coformulados es una opción en pacientes con deseos de disminuir el número de pastillas diarias, alteraciones gastrointestinales o dislipidemia (A-I).
Fundamento. Tres ensayos clínicos han demostrado que el cambio de IP/r a RAL es seguro virológicamente si los 2 ITIAN son completamente activos. El cambio se asocia en una mejoría de los niveles de CT, cLDL, cociente CT/cHDL y TG107,108. Los resultados del estudio SPIRAL108 sugieren que si el tiempo de supresión viral es muy prolongado, el riesgo de FV es menor, independientemente de la actividad de los ITIAN.
Recomendación- –
El cambio a 2 ITIAN activos más RAL es una opción para pacientes con dislipidemia que reciben TAR con 2 ITIAN más un IP/r (A-I).
Fundamento. Un ensayo clínico aleatorizado y abierto con pacientes con CVP suprimida y sin historia de FV ha demostrado que el cambio de TDF/FTC más un IP/r (fundamentalmente ATV/r, DRV/r o LPV/r) a TDF/FTC/COBI/EVG coformulados es mejor virológicamente. Los pacientes que cambiaron de tratamiento mejoraron significativamente los resultados en escalas de diarrea y plenitud abdominal; sin embargo, se reportaron más náuseas. Hubo una ligera mejoría en los niveles lipídicos, fundamentalmente en los pacientes que discontinuaron LPV/r109.
Recomendación- –
El cambio de TDF/FTC+ATV/r o DRV/r o LPV/r a TDF/FTC/COBI/EVG es virológicamente seguro. Este cambio es una opción para los pacientes que quieran simplificar su régimen actual y puede mejorar en algunos pacientes los síntomas digestivos asociados a ritonavir (A-I).
Fundamento. Dos ensayos clínicos aleatorizados y abiertos han demostrado que el cambio de 2 ITIAN+ATV/r a terapia dual con 3TC+ATV/r110 o de 2 ITIAN y LPV/r a terapia dual con 3TC+LPV/r111 no es inferior a la triple terapia.
Recomendación- –
El cambio de 2 ITIAN+ATV/r o LPV/r a terapia dual con 3TC+ATV/r o 3TC+LPV/r es una opción si el clínico quiere evitar o prevenir los efectos adversos causados por los ITIAN. Esta opción requiere que el paciente cumpla los siguientes criterios: a)ausencia de hepatitis crónica B; b)CVP<50 copias/ml durante al menos 6 meses, y c)ausencia de MR en el gen de la proteasa o fracasos virológicos previos a IP/r o 3TC (A-I).
Fundamento. La monoterapia con DRV/r QD112 o LPV/r BID113,114 no ha demostrado a largo plazo la noinferioridad frente a la terapia triple en los análisis por intención de tratar (ITT) si se considera el cambio de terapia aleatorizada igual a fracaso. La noinferioridad sí se ha demostrado en los análisis por ITT pura (ignorando los cambios de tratamiento, fundamentalmente las reinducciones con ITIAN). No existe acuerdo sobre cuál de estos análisis es más relevante clínicamente.
En los ensayos clínicos de monoterapia con DRV/r o LPV/r con seguimiento virológico adecuado no se ha demostrado un incremento en el riesgo de selección de MR en la proteasa114,115. En pacientes tratados con monoterapia tampoco se ha demostrado un aumento del riesgo de deterioro neurocognitivo asociado al VIH-1114,116, replicación viral discordante en el líquido cefalorraquídeo (LCR), aumento de los biomarcadores de inflamación117 o incremento del ADN celular del VIH-1 integrado118.
Comparada con el TAR triple, la monoterapia con DRV/r o LPV/r no ha demostrado beneficios a largo plazo aparte del ahorro económico. Sin embargo, tampoco hay datos empíricos que justifiquen que si un paciente es capaz de mantener la supresión virológica con DRV/r o LPV/r en monoterapia sea necesario utilizar además 2 ITIAN.
Este panel considera que no existe evidencia suficiente para recomendar el cambio proactivo a monoterapia con DRV/r o LPV/r en los pacientes que cumplan los criterios para el uso de esta estrategia. Sin embargo el panel considera que tampoco existen evidencias para oponerse al uso de monoterapia con DRV/r o LPV/r si el clínico quiere evitar o prevenir los efectos adversos causados por los ITIAN. Los factores que predicen éxito de la monoterapia son: adherencia elevada, supresión virológica prolongada y profunda119 y cifra nadir de linfocitos CD4+ mayor de 100 células/μl120.
La monoterapia con ATV/r o la combinación ATV/r y RAL no se recomienda debido a los peores resultados obtenidos en ensayos clínicos121-123.
Recomendación- –
Este comité considera que la monoterapia con DRV/r QD o con LPV/r BID es una opción si el clínico quiere evitar o prevenir los efectos adversos causados por los ITIAN y el paciente cumple los siguientes criterios: a)ausencia de hepatitis crónica B; b)CVP menor de 50 copias/ml durante al menos 6 meses, y c)ausencia de MR en el gen de la proteasa o FV previo a IP/r (B-I).
Tras el cambio de un régimen de TAR, el clínico debe evaluar en un plazo de 3-6 semanas el mantenimiento de la supresión virológica y las determinaciones de laboratorio pertinentes que dependerán del motivo del cambio (lipidograma, función renal, etc.). Una vez demostrada la continuación de la supresión virológica, el paciente puede volver a la rutina de visitas cada 4 o 6 meses.
Fracaso del tratamiento antirretroviralDefiniciones- –
Fracaso virológico: 2 determinaciones consecutivas de CVP >50copias/ml, a las 24 semanas del inicio del TAR. El FV puede ocurrir con o sin selección de MR. Si la CVP basal es elevada, pueden precisarse más de 24 semanas para alcanzar la CVP<50copias/ml, particularmente en regímenes sin INI.
- –
Repuntes virológicos transitorios (blips): valores de CVP aislados y transitorios entre 50 y 200 copias/ml. Los blips no se asocian a mayor riesgo de FV, aunque algunos autores lo han reportado así y con aparición de MR cuando son frecuentes124. En presencia de blips se recomienda evaluar la adherencia y la barrera genética del TAR frente a la resistencia.
- –
Fracaso inmunológico: incapacidad de obtener un recuento adecuado de linfocitos CD4+ a pesar de mantener una CVP<50 copias/ml. No se recomienda modificar el TAR, salvo que incluya FAR o combinaciones como ZDV o TDF+ddI.
En los estudios de registro de las pautas preferentes del TAR de inicio las tasas de FV a las 48 semanas oscilan entre el 4 y el 12% (véase el apartado «Tratamiento antirretroviral inicial»). En pacientes que inician TDF/FTC+ITINN o IP/r, la incidencia de FV en estudios de cohortes españoles es del 13%125. No existen datos de incidencia o prevalencia de FV en pacientes expuestos a 2 o más regímenes de TAR diferentes.
Los factores que influyen en el fracaso terapéutico son: a)dependientes del paciente: adherencia al TAR, dificultad de acceso o seguimiento; b)dependientes de los fármacos: errores de dosificación, potencia del TAR en pacientes con CVP elevada, concentraciones plasmáticas inadecuadas o interacciones medicamentosas, y c)dependientes del virus: preexistencia de MR transmitidas o adquiridas.
Objetivo del tratamiento tras el fracaso virológicoEl objetivo del nuevo TAR es conseguir de nuevo supresión viral mantenida (CVP<50 copias/ml). Para ello, debe instaurarse un TAR con 3, o al menos 2, FAR activos, incluyendo preferentemente al menos uno de una nueva familia. El TAR de rescate no debe retrasarse para evitar la acumulación de MR y la elevación de la CVP.
Estrategias para mejorar el éxito de los tratamientos de rescate• Facilitar la adherencia y tolerabilidad al TAR. Identificar causas de mala adherencia e intentar corregirlas; pautar un TAR cómodo, bien tolerado y de escasa toxicidad, valorando las posibles comorbilidades del paciente.
• Pruebas de resistencia. Los test de resistencia mejoran la eficacia del TAR de rescate126, especialmente cuando la prueba se realiza mientras el paciente recibe el TAR que ha fracasado. Cuando se dispone de estudios genotípicos previos, deben valorarse todas las MR acumuladas en los sucesivos FV. Para algunos IP/r y ETR se ha elaborado un índice ponderado de resistencia genotípica que cuantifica el peso de cada MR y establece un índice de respuesta al fármaco127. Los test ultrasensibles de secuenciación genotípica detectan poblaciones virales resistentes minoritarias y en algunos casos podrían mejorar el resultado del TAR de rescate128. La ausencia de MR durante un FV orienta a falta de adherencia o a realización inadecuada del test genotípico (>4 semanas sin TAR).
• Tropismo viral. Debe determinarse en cada FV, excepto si se sabe que existen cepas no-R5.
• Revisar el historial terapéutico. Identificar FAR que no se toleraron o regímenes de TAR que fracasaron con fármacos de barrera genética baja que hubieran podido seleccionar MR no detectadas en el test genotípico.
• Monitorización de concentraciones plasmáticas de fármacos. No se ha demostrado que mejore la eficacia del TAR de rescate. Debe reservarse para situaciones especiales, incluyendo la sospecha de mala adherencia.
Escenarios clínicos de fracaso virológicoFracaso virológico con viremias bajasBajo este epígrafe se consideran 2 situaciones:
- 1)
CVP de 50-200 copias/ml. En esta situación la CVP puede volver a quedar indetectable o mantenerse en niveles bajos sin necesidad de cambiar el TAR. Los test que utilizan técnicas de PCR en tiempo real (TaqMan® o Abbott RealTime®) presentan mayor sensibilidad para detectar viremias bajas (50-250 copias/ml)129,130. Si la CVP se mantiene entre 50-200 copias, la probabilidad de amplificar el ARN viral y detectar MR es muy baja131. Los pacientes suelen mantener recuentos de linfocitos CD4+ estables y no presentan progresión clínica. Algunos estudios han demostrado selección de nuevas MR132, FV133 y asociación a translocación bacteriana e inflamación sistémica134. En general, no se recomienda modificar el TAR, aunque el régimen debe ser de alta barrera genética.
- 2)
CVP: 200-1000 copias/ml. Este grado de viremia se asocia a selección de MR frente a ITIAN, ITINN e INI130. Debe realizarse un test de resistencias que son factibles a partir de CVP>200 copias/ml. Si el historial terapéutico o el test genotípico permiten administrar un TAR potencialmente eficaz, debe considerarse cambiar el TAR.
En ambas situaciones está contraindicada la intensificación terapéutica añadiendo un solo FAR activo. Debe valorarse la potencia del TAR, la adherencia, errores de prescripción y posibles interacciones medicamentosas o alimentarias.
Fracaso virológico precozSe habla de FV precoz cuando fracasa la primera línea de TAR. La selección de MR y las pautas de segunda línea difieren dependiendo de la pauta de inicio utilizada:
• Fracaso virológico a 2 ITIAN + ITINN. Las MR seleccionadas con mayor frecuencia tras el FV con EFV o NVP son: K103N, L100I o Y181C. La K103N aislada mantiene sensibilidad a RPV y ETR. El FV con RPV selecciona las E138K e Y181C, que generan resistencia a todos los ITINN. El FV a ITINN puede acompañarse de MR a ITIAN, especialmente la M184V, y con menor frecuencia la K65R.
• Fracaso virológico a 2 ITIAN + IP/r. La probabilidad de seleccionar MR a IP es muy baja. Los IP/r protegen de la selección de MR a ITIAN, siendo infrecuentes y limitándose solo a la M184V.
• Fracaso virológico a 2 ITIAN + INI. El FV a RAL o EVG selecciona MR cruzadas entre ambos INI: T66K, E92Q, Q148H/K/R y N155H, que con frecuencia se acompañan de MR a ITIAN. Los ensayos clínicos en pacientes naïve han demostrado que DTG es un INI de alta barrera genética frente a la resistencia, siendo excepcional detectar MR en el gen de la integrasa o de la transcriptasa inversa tras un FV con DTG.
Existen pocos ensayos clínicos aleatorizados que hayan evaluado la eficacia de diferentes combinaciones de FAR en pacientes con FV a alguno de los TAR de primera línea (tabla 7). En general, se recomienda la utilización de un IP/r con otros 2 FAR, preferentemente ITIAN, que conserven la actividad antiviral.
Principales ensayos clínicos de tratamiento antirretroviral en pacientes con fracaso virológico precoz y avanzado
| Fármaco (ensayo clínico) | Criterios de inclusión | Diseño | Brazos de comparación | n | Endpoints48 semanas | Principales conclusiones | Otros resultados |
|---|---|---|---|---|---|---|---|
| Estudios de rescate precoz | |||||||
| DRV/r (ODIN) | • CV>1.000 copias• CD4+>50 células• No MR a DRV | Fase IIIbNo inferioridad(Δ:−12%)ITT-TLOVR | • DRV/r 800/100 QD• DRV/r 600/100 BID(ambos+ITIAN) | 294296 | • 72,1%<50 copias• 70,9%<50 copias• Δ 1,2% (IC 95% −6,1 a 8,5%) | DRV/r QD es no inferior a DRV/r BID en pacientes con FV sin mutaciones a DRV | • Los EA inferiores en pauta QD (7,8%) frente a BID (15,2%) y con mejor perfil lipídico |
| LPV/r mono (HIVSTAR) | • TAR: 2 ITIAN+ITNN• CV ≥ 1 000 copias• IP naïve | Fase IVAleatorizadoAbiertoNo inferioridad ITT | • LPV/r (monoterapia)• LPV/r+TDF+3TC | 9897 | • 61%<50 copias• 83%<50 copiasITT p<0,01 | • LPV/r mono es menos eficaz que LPV/r+TDF+3TC como terapia de 2.a línea | • CV> 5 u Log y presencia de TAM se asociaron a más fracaso con LPVr-monoterapia |
| LPV/r+RAL (SECOND-LINE) | • CV>500 copias• 1.er TAR ≥ 24 semanas• Nunca LPV/r | Fase IIIb/4AleatorizadoAbiertoNo inferioridad(Δ:−12%) en ITT | • LPV/r+RAL• LPV/r+2 ITIAN | 271270 | • 83%<200 copias 48 semana• 81%<200 copias 48 semanaΔ: 1,8% (IC 95% −4,7 a 8,3) | • LPV/r+RAL es no inferior al estándar de tratamiento LPV/r+2 ITIAN | • LPV/r+RAL es una terapia bien tolerada y segura• Permite ahorrar ITIAN• No precisa realizar test genotípico |
| ETR (TMC125-C227) | • CV>1.000 copias/ml• FV a 1.er TAR con EFV o NVP• Resistencia a ITINN e ITIAN• Naïve a IP | Fase IIAleatorizadoAbierto | • ETR 800mg BID• IP o IP/rAmbos con 2 ITIAN a criterio del investigador | 5957 | • Descenso CV-VIH (log10) desde valor basal a semana 12 y 24:• ETR: −1,39 y −1,51 log10 copias/ml• IP: −2,16 y −2,13 log10 copias/ml | • ETR es inferior a IP, ambos con 2 ITIAN en pacientes con FV y MR a ITINN e ITIAN• La presencia de MR basales fue la principal razón del FV | • Diferencia (ETR vs DRV/r) porcentaje de sujetos con CV<50 copias/ml en semana 12 fue −27.8% (IC 95%: −46,8%, −8,8%) |
| Estudios de rescate avanzado | |||||||
| DRV/r (TITAN) | • CV>1.000 copias• TAR ≥ 12 semanas• Nunca LPV/r, TVR, DRV o ENF | Fase IIIAleatorizadoNo ciegoNo inferioridad(Δ:−12%) ITT | • DRV/r BID+TO• LPV/r BID+TO | 298297 | • 71%<50 copias (77%<400 copias)• 60%<50 copias (68%<400 copias)Δ 11%(IC 95%: 3-19)<50 copias Δ 9% (IC 95%: 2-16)<400 copias | DRV/r no inferior LPV/r y cumple criterios de superioridad para CV< 50 copias y 400 copias | • DRV/r>LPV/r si CV> 5uLog• FV 10% (DRV), 22% (LPV)• MR a IP e ITIAN más frecuentes con LPV |
| DRV/r (POWER I/II) | • CV>1.000 copias• Multifracaso• MR a IP ≥ 1 | Fase IIBAleatorizadoITT-LOVR | • DRV/r BID 600/100+TO• IP/r comparador+TO | 131124 | • 45%<50 copias• 10%<50 copiasΔ 37% (IC 95%: 25-46)<50 copias | DRV/r 600/100 BID>IP/r comparador | • La superioridad de DRV/r fue independiente del uso de ENF, CV basal, número de mut. a IP o fármacos activos en la TO |
| RAL (BENCHMRK I/II) | • CV>1.000 copias, mientras reciben TAR• Resistencia documentada al menos a un fármaco de las 3 familias: ITIAN, ITINN e IP | Fase IIIAleatorizadoDoble ciego | • RAL+TO• Placebo+TO | 462237 | • 62,1%<50 copias semana 48• 32,9%<50 copias semana 48p<0,001 | En pacientes VIH con opciones limitadas, RAL+TO es superior a placebo+TO | • Cuando RAL se asoció a DRV/r+ENF la eficacia fue del 98%• La seguridad de RAL fue similar a placebo• RAL presenta una barrera genética baja |
| ETV (DUET I/II) | •>8 semanas con TAR• CV>5.000 copias• ≥ 1 MR ITIAN• ≥ 3 MR IP | Fase IIIAleatorizadoDoble ciego | • DRV/r+ETV• DRV/r+placebo(ambos+ITIAN) | 599604 | • 61%,<50 copias• 40%,<50 copiasp<0,0001 | • ETV es superior a placebo en pacientes con FV y experiencia a varios TAR | • 91% de los pacientes con CV<50 copias semana 48 persisten en la semana 96 con<50 copias• Los eventos de sida0 y muerte disminuyeron en pacientes con ETV+ENF |
| MVC (MOTIVATE I/II) | • CV>5.000 copias• Tropismo R5• FV a ITIAN, ITINN e IP | Fase IIIAleatorizadoDoble ciego | • MVC QD+TO• MVC BID+TO• Placebo+TO | 414426209 | CV<50 copias: 43,2%CV<50 copias: 45,5%CV<50 copias: 16,7%p<0,001 | MRV es superior a placebo en pacientes con tropismo R5 | • CV<50 copias: 64% (TO+ENF)• CV<50 copias: 61% (TO+ENF)• CV< 50 copias: 27% (TO+ENF) |
| DRV/RAL/ETV (TRIO) | • CV>1.000 copias• Naïve a DRV/ETV/RAL• FV durante TAR• ≥ 3 mut. proteasa (sensible DRV)• ≥ 3 mut. ITIAN• Susceptible a ETV | Fase IIAbiertoNo comparativo | • DRV/r 600/100 BID+RAL 400mg BID+ETV 200mg BID | 103 | • CV<50 copias semana 24: 95% (IC 95%: 85-96%)• CV<50 copias semana 48: 86%(IC 95%: 80-93%) | DRV/r+ETV+RAL es muy eficaz en pacientes con pocas opciones | • La mediana de CD4+ se incrementó en 108 células/mm3• Incidencia EA grado 3/4: 14,6% |
| EVG/r | • CV>1.000 copias• MR o más de 6 meses≥>2 clases de FARV | Fase IIIDoble ciegoAleatorizadoNo inferioridadΔ: −10% en ITT | • EVG 150mg (85mg si LPV/r o ATV/r)• RAL 400mg/BIDAmbas pautas con IP/r y otro FAR activo | 361363 | • CV<50 copias: 59%• CV<50 copias: 58%Δ: 1,1% (IC 95%: −6,0 a 8,2) | EVG en combinación es no inferior a RAL ambos con un IP/r | • La administración de EVG es QD.No diferencias de efectos adversos entre ambos brazos |
| DTG (SAILING) | • CV ≥ 1.000 copias/ml• Resistencia ≥ 2 familias de FAR• 1 o 2 FAR activos en la TO | • Fase III• Aleatorizado• Doble ciego• No inferioridad (Δ-12%) en ITT• Análisis superioridad | • DTG 50mg QD+TO• RAL 400mg BID+TO | 354361 | • 71%<50 copias semana 48• 64%<50 copias semana 48Δ: 7,4% (IC 95%: 0,7 a 14,2%) en ITT | DTG 50mg QD es superior a RAL BID ambos con TO en pacientes con FV y experiencia previa a TAR | • Los pacientes con DTG presentaron menos FV y menos mutaciones de resistencia a INI• Los efectos adversos fueron similares en ambos grupos |
| DTG(VIKING I-II) | • CV ≥ 1.000 copias/ml• Resistencia genotípica a RAL• Resistencia a ITIAN, ITNN, IF o IP• ≥1 FAR activo en TO de la cohorte 1 | • Fase IIb• 2 cohortes• 1.a fase monoterapia funcional: DTG• 2.a fase DTG+TO | • DTG 50mg BID+TO• DTG 50mg QD+TO | 2427 | • 75%<50 copias 24 semamas• 41%<50 copias 24 semanas | DTG 50mg. BID+TO es más eficaz que DTG QD en pacientes con resistencia a RAL | • La toxicidad fue similar en ambos grupos• Selección resistencias a InInt: <15% de todos los pacientes• No se detectaron nuevas mutaciones a InInt |
| DTG (VIKING-III) | • CV ≥ 500 copias/ml• Resistencia genotípica a RAL/EVG• Resistencia a ≥ 2 familias de FAR• ≥ 1 FAR activo en TO | • Fase III• Abierto, brazo único | DTG 50mg BID+TO | 183 | • 69%<50 copias/ml 24 semanas | DTG 50mg BID + TO es eficaz en pacientes con fracaso previo a RAL o EVG y otros FAR | • La presencia de MR basales: Q148+ ≥ 2 mutaciones a InInt reduce la eficacia de DTG• La tasa de suspensión por efectos adversos con DTG 50mg BID es baja (3%) |
DRV/r es el IP/r de elección en cualquiera de las líneas de rescate analizadas. El estudio ODIN135 demostró que DRV/r QD (800/100)+2 ITIAN era no inferior a DRV/r (600/100) BID en pacientes con FV y sin MR a DRV. El estudio TITAN136 demostró que en pacientes con exposición limitada a FAR (experiencia previa a ITIAN, ITINN e IP) pero naïve a LPV/r, DRV/r 600/100 BID es superior a LPV/r, ambos con terapia optimizada (TO).
En pacientes que fracasan a un primer TAR basado en ITINN (NVP, EFV), LPV/r+TDF/FTC es claramente superior a LPV/r en monoterapia (estudio HIV-STAR)137. En el mismo escenario, una pauta de biterapia (LPV/r+RAL) resulta no inferior a LPV/r+2 o 3 ITIAN (estudio SECOND-LINE)138. Esta pauta puede ser una opción en pacientes que no pueden recibir TDF o ABC.
Fracaso virológico avanzadoEn este escenario, la mayoría de los pacientes han experimentado FV con las 3 familias de FAR más utilizadas: ITIAN, ITINN e IP, y pueden detectarse MR a las 3 familias.
Varios ensayos clínicos han evaluado diferentes pautas de TAR de rescate (tabla 7). Estos estudios no son comparables entre sí por la heterogeneidad de la población, tratamientos previos, criterios de eficacia, tiempo de seguimiento y TO utilizada. Tanto ENF, como TPV/r, DRV/r, ETR, MVC y RAL, han demostrado superioridad frente a placebo en combinación con la TO disponible cuando se realizaron los estudios.
DRV/r es superior a otros IP/r en este escenario (estudios POWER139 y TITAN136). Cuando existe alguna MR mayor a DRV, pero el virus es sensible al fármaco, se recomienda utilizar DRV/r 600/100mg BID; en ausencia de MR es suficiente DRV/r 800/100mg QD140. La combinación DRV/r+ETR+TO es significativamente superior a DRV/r+TO en pacientes con ≥1MR a ITINN y ≥3 mutaciones a IP (estudios DUET)141.
En cualquier línea de rescate, añadir un FAR de una nueva familia aumenta de forma significativa la eficacia del TAR. Así, en pacientes con CVP>5.000 copias/ml, tropismo R5 y resistencia a FAR de 3 familias (ITIAN, ITINN e IP), MVC, en pauta BID o QD, +1IP/r +TO mejoró la eficacia del TAR (estudios MOTIVATE)142. RAL+TO en pacientes con CVP>1.000 copias/ml, con MR al menos a un FAR de cada una de las 3 familias, aumentó significativamente la proporción de pacientes que alcanzaron CVP<50 copias/ml a las 48 semanas (estudio BENCHMRK)143. El estudio TRIO144, abierto y no comparativo, evaluó la eficacia y la seguridad de un TAR que contenía DRV/r+ETR+RAL. Incluyó pacientes con CVP>1.000 copias/ml e infección por VIH-1 multirresistente. En la semana 48, 89 pacientes (86%; IC95%: 80 a 93%) lograron una CVP<50 copias/ml. EVG se comparó con RAL, ambos con TO (IP/r+1FAR activo), en pacientes con CVP>1.000 copias/ml y experiencia o resistencia a ≥2 familias de FAR. EVG resultó no inferior a RAL en la semana 48 (estudio GS-US-183-0145)145.
DTG 50mg QD resulta superior a RAL, ambos con TO, en pacientes naïve a INI, con CVP>1.000 copias/ml y resistencia a ≥2 familias de FAR (estudio SAILING)146. El estudio VIKING147 analizó la eficacia de DTG 50mg QD (cohortei) y 50mg BID (cohorteii) en 51 pacientes con resistencia previa a RAL. En la semana 24, el 41% de los pacientes de la cohortei tenían CVP<50 copias/ml y el 75% en la cohorteii. El estudio VIKING-3148, un ensayo clínico abierto y no aleatorizado, analizó la eficacia de DTG 50mg BID en 183 pacientes con fracaso previo a RAL o EVG. El 69% de los pacientes consiguieron una CVP<50 copias/ml en la semana 24. La presencia de la mutación Q148H/R en el gen de la integrasa, más otras adicionales, se relacionó con una menor eficacia de DTG.
TPV y ENF presentan alta toxicidad, y su uso solo se aconseja cuando no es posible conformar un tratamiento óptimo con otros FAR activos.
Fracaso virológico en pacientes sin opciones terapéuticasEn este escenario resulta imposible diseñar un TAR con al menos 2FAR plenamente activos. El FV en el paciente multitratado no siempre conduce de forma rápida a inmunodeficiencia y progresión clínica. La mayoría de pacientes continúan con recuentos de CD4+ relativamente estables, especialmente si continúan con TAR y la CVP se mantiene entre 10.000-20.000 copias/ml. En esta situación se recomienda: a)remitir al paciente a un centro especializado con experiencia y acceso a nuevos FAR mediante ensayos o accesos expandidos; b)mantener un TAR no supresor que sea cómodo, poco tóxico y que disminuya la capacidad replicativa viral y no acumule MR que comprometan la respuesta a futuros FAR; se recomienda el uso de 2 o 3 ITIAN, que incluyan 3TC o FTC, y simultáneamente AZT o TDF; 3TC y FTC inducen la mutación M184V que compromete la capacidad replicativa del VIH-1 y puede resultar útil en esta situación149; en cuanto sea posible, este tratamiento debe cambiarse a un TAR supresor con 2-3 FAR activos, y c)no debe interrumpirse el TAR, ya que el descenso de linfocitos CD4+ es mayor que si se continúa con un TAR no supresor.
Recomendaciones- –
El objetivo del TAR de rescate es conseguir una CVP<50 copias/ml (A-II).
- –
El cambio del TAR motivado por FV debe efectuarse precozmente para evitar la acumulación de mutaciones y facilitar la respuesta al nuevo tratamiento (A-III).
- –
El nuevo TAR debe contener 3 FAR totalmente activos y al menos uno de una nueva familia. Si no es posible, se recomienda la combinación de 2 FAR plenamente activos y otros que conserven actividad virológica parcial (A-I).
- –
Se debe realizar un estudio de resistencia y de tropismo viral para confeccionar el mejor régimen alternativo. La prueba debe realizarse mientras el paciente está recibiendo el TAR que ha fallado o lo antes posible tras la suspensión. Si se dispone de pruebas genotípicas previas, deben valorarse todas las MR detectadas (A-I).
- –
Deben analizarse las causas que motivaron el FV: adherencia, interacciones medicamentosas o alimentarias, historia farmacológica y toxicidades previas. El nuevo TAR debe ser lo más cómodo y mejor tolerado posible (A-III).
- –
En pacientes con FV, DRV/r es el IP/r que ha demostrado mayor eficacia en todas las líneas de rescate. En presencia de MR mayores a DRV se recomienda en dosis de 600/100mg BID (A-I).
- –
DTG es el INI de elección en pacientes con FV y naïve a INI (A-I). Si ha habido fracaso previo a RAL o EVG, la dosis de DTG debe ser 50mg BID acompañado de TO (A-II).
- –
El uso de TPV/r, ENF o análogos de la timidina queda restringido a pacientes sin otras posibilidades terapéuticas (A-III).
- –
En general, no se recomienda modificar el TAR en pacientes con FV de bajo grado (50-200 copias/ml) y que reciben un régimen de TAR de alta barrera genética frente a la resistencia. En pacientes con viremias superiores (>200 copias/ml) se recomienda realizar un test genotípico en un laboratorio de referencia y pautar un nuevo régimen de TAR en base a las MR o al historial terapéutico del paciente. No se recomienda la intensificación del TAR con un solo fármaco (A-III).
- –
No se recomienda suspender el TAR en pacientes con FV avanzado y sin opciones terapéuticas. En esta situación se recomienda usar FAR que disminuyan la capacidad replicativa viral y que no seleccionen nuevas MR que comprometan futuros tratamientos (A-III).
- –
En pacientes sin posibilidades terapéuticas se recomienda vigilar la evolución del recuento de linfocitos CD4+ y de la CVP, y consultar con clínicos y virólogos con experiencia en resistencia, tratamientos de rescate y con acceso a FAR de uso restringido (B-III).
Se entiende por adherencia al TAR la capacidad del paciente para implicarse correctamente en la elección, inicio y cumplimiento del mismo, a fin de conseguir una supresión adecuada de la replicación viral. La adherencia incorrecta es la primera causa de fracaso terapéutico. Entre los factores asociados con una adherencia incorrecta destacan: mala relación médico-paciente, consumo de drogas, enfermedad mental, deterioro neurocognitivo, bajo nivel educativo, barrera idiomática, falta de apoyo social y complejidad y efectos secundarios del TAR150-153. Por el contrario, el apoyo emocional, la capacidad para incluir la medicación en las actividades de la vida diaria y la comprensión de la importancia de la adherencia son factores que predicen una adherencia correcta. La adherencia describe la calidad en la ejecución del tratamiento prescrito; aspectos vinculados, como el acceso y el mantenimiento del TAR, son esenciales para el éxito del mismo154.
Antes de iniciar el TAR conviene preparar al paciente, identificar las situaciones que puedan dificultar la adherencia e intentar corregirlas. Durante la realización del TAR es fundamental evaluar periódicamente la adherencia. Se recomienda utilizar más de un método, como la entrevista, los cuestionarios estructurados, el recuento de medicación sobrante y el registro de dispensación por farmacia.
Cuando existen niveles subterapéuticos de FAR, el VIH puede replicarse y desarrollar resistencias. Debe destacar que no solo es importante el porcentaje de dosis omitidas, sino también los patrones de adhesión subóptima: las interrupciones del TAR tienen una mayor repercusión en la respuesta virológica que la omisión ocasional de dosis.
El grado de adherencia necesario para lograr el éxito terapéutico no se conoce con certeza. La relación entre la adherencia al TAR, el control virológico y el desarrollo de resistencia varía entre las diferentes clases de FAR y la situación clínica del paciente (CVP, duración de la indetectabilidad de la CVP). Así, con los IP/r la aparición de resistencias es mucho más difícil con cualquier nivel de adherencia debido a su elevada barrera genética frente a la resistencia155. El INI DTG, también tiene una alta barrera genética, pero la experiencia en la práctica clínica es aún escasa. Por tanto, ante un paciente en el que se prevé una adhesión incorrecta al TAR, es mejor iniciar un régimen de TAR basado en IP/r (o DTG), que evitan el riesgo de seleccionar MR en caso de incumplimiento.
Si se detecta falta de adherencia debe intervenirse de forma activa para corregirla. En ocasiones los pacientes presentan falta de adherencia selectiva, es decir, solo a alguno de los componentes del TAR, y esta falta de adherencia se relaciona con el desarrollo de FV156. La coformulación de FAR simplifica el TAR y puede prevenir este problema, mejorando la adherencia global157. En este sentido, el uso de regímenes completos en comprimido único constituye la estrategia más eficiente para prevenir la mala adherencia selectiva de fármacos. Además de mejorar la adherencia, el uso de regímenes en comprimido único se ha asociado con menores tasas de hospitalización y de costes en cuidados médicos158.
Las estrategias para mejorar la adherencia son múltiples. Probablemente la intervención que ha demostrado una mayor eficacia ha sido el soporte interpersonal estructurado, en el que personal sanitario entrenado emplea estrategias individualizadas159.
El tratamiento directamente observado (TDO) ha demostrado, en un metaanálisis, un discreto aumento en la probabilidad de conseguir CVP indetectable, mayor incremento de linfocitos CD4+ y adherencia superior al 95%, mientras se mantiene el TDO.
GeSIDA y el PNS, conjuntamente con la Sociedad Española de Farmacia Hospitalaria, han revisado los factores que influyen en la adherencia, los métodos de evaluación y las posibles estrategias de intervención160. Remitimos a los lectores a este documento de consenso (http://www.msssi.gob.es/ciudadanos/enfLesiones/enfTransmisibles/sida/docs/recomendacionesAdherenciaTrtoAntirretroviral062008.pdf) para profundizar en el tema de la adherencia al TAR.
Recomendaciones- –
Antes de iniciar el TAR se debe preparar al paciente e identificar y corregir las causas potenciales de adherencia incorrecta (A-III).
- –
Una vez iniciado el TAR, se recomienda efectuar un control a las 2-4 semanas para comprobar la adherencia y eventualmente corregirla (A-III).
- –
La adherencia debe monitorizarse y reforzarse coincidiendo con las visitas clínicas (A-III).
- –
El control de la adherencia debe realizarse por un equipo multidisciplinar, adaptado a la disponibilidad de cada centro, que incluya médicos, personal de enfermería, profesionales de apoyo psicológico y de farmacia hospitalaria (A-III).
- –
En pacientes con cumplimiento irregular es preferible utilizar pautas basadas en IP/r (y probablemente DTG) para prevenir la selección de resistencias (A-III). A pesar de la poca experiencia clínica disponible, los datos iniciales parecen apoyar que las pautas basadas en DTG pueden ser también útiles en este tipo de pacientes (B-III).
- –
La combinación a dosis fijas de FAR simplifica el TAR y, por tanto, facilita el cumplimiento mantenido. El uso de regímenes completos en comprimido único constituye la estrategia más eficiente para prevenir la mala adherencia selectiva de fármacos (A-II).
La tolerabilidad depende de aspectos relacionados con la toma del FAR (número y tamaño de los comprimidos, requisitos de administración, incidencia e intensidad de efectos secundarios inmediatos), pero también de factores del paciente (edad, sexo, peso, situación clínica y expectativas respecto al TAR). En el último decenio, tanto los FAR como su galénica han mejorado notablemente, lo cual ha favorecido su tolerabilidad y su aceptación por los pacientes.
Clasificación cronológica de los efectos adversosLos efectos adversos de los FAR pueden ser inmediatos (a corto plazo) o tardíos (a largo plazo). Los efectos inmediatos se producen en los primeros días o semanas de tratamiento, mientras que los tardíos aparecen al cabo de meses o años después del inicio de este.
Efectos adversos inmediatosEstán bien definidos, en algunos casos pueden preverse y suelen ser fáciles de controlar. Afectan principalmente a la esfera digestiva, cutánea o neuropsicológica, y su incidencia y factores asociados son conocidos.
Entre los FAR actualmente recomendados, los IP/r pueden producir efectos digestivos66,161; los ITINN de primera generación (particularmente NVP) hepáticos y RHS162; ABC produce RHS en pacientes con HLA-B*5701 positivo28; DRV, exantema161, y EFV puede producir efectos neuropsicológicos162.
Recomendaciones- –
Se debe evitar el uso de FAR cuyos efectos adversos inmediatos sean similares a manifestaciones clínicas o alteraciones de laboratorio ya presentes en un determinado paciente (A-II). La determinación del alelo HLA-B*5701 es obligada antes de prescribir ABC, ya que tiene un valor predictivo positivo de casi el 100% para el riesgo de RHS a este fármaco (A-I).
- –
Se debe explicar al paciente cómo tomar correctamente una pauta de TAR y la posibilidad de que ocurran determinados efectos adversos inmediatos. Al iniciar un régimen de TAR se debe explicar qué actitud debe tomar el paciente si ocurre un determinado efecto adverso y, en cualquier caso, se debe facilitar siempre la posibilidad de comunicación directa con el médico. Los efectos adversos inmediatos leves se pueden tratar sintomáticamente, valorando la evolución de la tolerabilidad del paciente. Si el efecto tiene gran intensidad o duración prolongada, o no es asumible por el paciente, se debe cambiar el o los FAR potencialmente implicados (A-I).
Se conocen peor que los inmediatos y son más difíciles de prever y controlar. Potencian los síntomas de las enfermedades crónicas asociadas al envejecimiento y afectan al funcionamiento de órganos y sistemas. El perfil de órganos y sistemas al que pueden afectar los diversos FAR, así como los factores de riesgo asociados a tal afectación, no se conoce en su totalidad, particularmente en lo que concierne a los FAR más recientes. La tabla 8 resume los efectos secundarios tardíos más característicos de los FAR actuales66,163-180.
Efectos secundarios tardíos más característicos de los antirretrovirales más utilizados en la actualidad
| Fármacos | Efectos adversos |
|---|---|
| Tenofovir(Factores de riesgo: afectación renal u ósea previas, presencia de factores de riesgo convencionales, duración de la exposición a TDF y concomitancia de tratamiento con IP/r) | • Descenso del FGe. Puede aumentar el riesgo de insuficiencia renal• Disminución de la densidad mineral ósea. Puede aumentar el riesgo de osteoporosis y fracturas osteoporóticas• Puede producir hipofosfatemia por tubulopatía proximal renal y agravar un eventual déficit de vitamina D |
| Abacavir | • Se ha asociado a riesgo de infarto de miocardio en pacientes con riesgo cardiovascular elevado, aunque este efecto clínico es controvertido |
| Inhibidores de la proteasa | • Pueden aumentar el riesgo de enfermedad cardiovascular por su efecto lipídico y quizá por otros efectos que no se conocen bien• LPV/r ha sido asociado a un mayor riesgo de infarto de miocardio, ATV no ha sido asociado a infarto de miocardio, y sobre DRV/r no se dispone de información al respecto• ATV puede producir hiperbilirrubinemia y excepcionalmente ictericia y colelitiasis• LPV/r y ATV/r (no hay datos de DRV/r) se han asociado a un mayor riesgo de disminución del FGe, aunque este efecto clínico es controvertido y podría deberse a la interacción con TDF cuando se administran concomitantemente• ATV/r y DRV/r se han asociado a litiasis renal |
| Efavirenz | • Trastornos neuropsicológicos mantenidos (aunque sean de bajo grado pueden resultar difíciles de tolerar a largo plazo)• Se ha asociado a un mayor riesgo de deterioro neurocognitivo, aunque este efecto clínico es controvertido. En cultivos neuronales induce neurotoxicidad• Disminuye el nivel plasmático de 25-OH vitamina D, pero el significado clínico de esta alteración es desconocido• Se ha asociado a ginecomastia, aunque este efecto clínico es controvertido• Puede ser teratógeno y está contraindicado en mujeres que deseen quedarse embarazadas |
El peso relativo que suelen tener los FAR en la producción o desarrollo de enfermedades crónicas es en general pequeño y mucho menor que el de otros factores de riesgo clásicos, ya conocidos en la población general, que en algunos casos están sobrerrepresentados en pacientes con infección por el VIH, como son el consumo de tabaco, alcohol u otras drogas, una dieta inadecuada, o la ausencia de ejercicio físico.
En general, el riesgo absoluto de efectos secundarios tardíos con los FAR actualmente recomendados es muy pequeño, y el beneficio en términos de salud global de una pauta de TAR efectiva respecto a no tratar está fuera de duda. Sin embargo, en pacientes en alto riesgo o con enfermedades crónicas ya diagnosticadas, el efecto de determinados FAR puede contribuir por sí mismo a desencadenar o hacer progresar, respectivamente, tales enfermedades crónicas.
Recomendaciones- –
Se debe individualizar el TAR, evaluando el riesgo o la presencia de enfermedades crónicas, de manera que la pauta elegida no contenga FAR que puedan favorecer la aparición o progresión de las mismas (A-II).
- –
La retirada de algunos FAR implicados en efectos adversos tardíos puede mejorar, al menos parcialmente, la alteración clínica subyacente, aunque se desconoce si tal modificación puede alterar la historia natural de la enfermedad crónica en cuestión o la supervivencia. Los FAR contribuyen de forma colateral al riesgo o progresión de determinadas enfermedades crónicas, pero hay otros factores que generalmente son más importantes; se recomienda priorizar la intervención sobre dichos factores (A-II).
Un número significativo de pacientes con infección por el VIH reciben diversos medicamentos además de los FAR. Ocasionalmente toman productos de herboristería, suplementos dietéticos o medicinas alternativas. Las interacciones de los FAR entre sí o con otros medicamentos pueden tener una repercusión clínica importante. En un estudio con 1.497 pacientes de la cohorte suiza se estimó que el 35% de los pacientes menores de 50 años y el 51% de los mayores de esta edad podían presentar interacciones potencialmente importantes (asociaciones contraindicadas o que requerían ajuste de dosis que no había sido realizado)181.
Las interacciones más relevantes suelen ser las farmacocinéticas (dan lugar a una modificación de concentraciones), en especial las que afectan al metabolismo. Los FAR son sustratos de uno o varios sistemas enzimáticos, y a la vez pueden comportarse como inductores y/o inhibidores de los mismos. La inducción producirá una disminución de las concentraciones del otro fármaco (sustrato), pudiendo disminuir su eficacia. La inhibición ocasionará un aumento de las concentraciones con mayor riesgo de toxicidad. En general, la inducción es un proceso lento (días o semanas), mientras que la inhibición se produce rápidamente (horas). Algunos FAR pueden ser inhibidores e inductores al mismo tiempo, predominando uno u otro efecto.
El sistema metabólico más importante es el citocromo P450 (CYP) y su principal isoenzima (CYP3A4). Muchos FAR, especialmente los IP e ITINN, y muchos otros medicamentos que a menudo reciben los pacientes, son sustratos, inhibidores o inductores del CYP. La potente inhibición enzimática que producen ritonavir y COBI se utiliza para potenciar otros FAR. COBI, a diferencia de ritonavir, no presenta actividad antirretroviral.
Otra vía metabólica es la conjugación de los FAR o de sus metabolitos con otros productos, por ejemplo la glucuronidación mediante la enzima UDPGT. Diversos FAR son inductores o inhibidores de UDPGT. Ritonavir inhibe varias subfamilias del CYP y es inductor de UDPGT, mientras que ATV inhibe tanto CYP como UDPGT.
Ciertos transportadores, como la glucoproteína-P (P-gp), pueden alterar la biodisponibilidad de algunos FAR y su distribución por el organismo. Estos transportadores pueden ser también inducidos o inhibidos por diversos fármacos.
En la tabla 9 se detallan las características de los FAR como sustratos, inductores o inhibidores del CYP. Esta clasificación nos permite determinar posibles interacciones teóricas para una combinación determinada. Se indican también en ella las asociaciones contraindicadas. Los ITIAN tienen pocas interacciones metabólicas, por lo que no se han incluido.
Antirretrovirales como sustratos, inductores e inhibidores del citocromo P-450, glucuronidación (UGT), glucoproteína-P (P-gp) y otros transportadores. Asociaciones contraindicadas o no recomendadas
| Clase | Antirretroviral | Es sustrato de:a | Es inductor de:a | Es inhibidor de:a |
|---|---|---|---|---|
| ITINN | Efavirenz | CYP2B6, 3A4GlucuronidaciónUGT2B7 | CYP3A4CYP2C19 (in vivo; voluntarios sanos)CYP 2B6Glucuronidación(UGT1A1)Autoinduce su propio metabolismo | CYP2C9 y 2C19 (in vitro; sin embargo, in vivo ha habido informes contradictorios tanto de exposiciones aumentadas como disminuidas a sustratos de estas enzimas coadministrados con efavirenz. El efecto neto no está claro)3A4(in vitro)CYP2C8UGT1A9 y UGT1A4 (in vitro)P-gp |
| Asociaciones contraindicadas o no recomendadas con efavirenz | ||||
| Antirretrovirales:ATV/rb (considerar ATV/r 400/100mg c/24h en naïve; evitar en pretratados)DRV/r 800/100 mg/24h (emplear DRV/r 600/100mg/12h)ETRFPV no potenciado (emplear FPV/r 700/100mg/12h)LVP/r (considerar LVP/r 500/125mg/12h; 5 comp. de 100/25mg por toma)MVC: ↑MVC 600mg/12h (en ausencia de inhibidores potentes del CYP3A4; en presencia de los mismos 150 mg/12 de MVC)NVPRPVSQV no potenciado | VHC:Boceprevir contraindicado(Nota: con telaprevir permitido ↑ TVR a 1.125 mg/8h) | Otros:Anticonceptivos orales (emplear métodos de barrera)AmodiaquinaAtovacuonaCarbamacepinaClaritromicina (para tratamiento de MAC, valorar azitromicina)Derivados de ergotaminaGingko biloba (extracto)Hypericum (hierba de San Juan)ItraconazolKetoconazolMidazolamPimozidaPosaconazolProguaniloTerfenadinaTriazolamVoriconazol (evitar/ajustar dosis. Según datos farmacocinéticos: 400 mg/12h de voriconazol+300 mg/24h de EFV)Zumo de pomelo | ||
| Nevirapina | CYP3A4, 2B6, 2D6GlucuronidaciónP-gp | CYP3A4 (induce su propio metabolismo), 2B6P-gp | — | |
| Asociaciones contraindicadas o no recomendadas con nevirapina | ||||
| Antirretrovirales:ATV/rEFVETREVG/COBIFPV no potenciadoLPV/r (se recomienda LVP/r 500/125 mg/12h; 5 comp. de 100/25mg por toma)RPVSQV no potenciado | VHC:Boceprevir y telaprevir (por ausencia de datos) | Otros:Anticonceptivos orales (emplear métodos de barrera o medroxiprogesterona depot)Claritromicina (para tratamiento de MAC, valorar azitromicina)Hypericum (hierba de San Juan)KetoconazolItraconazolRifampicinaVoriconazol | ||
| Etravirina | CYP3A4, CYP2C9/19CYP2C18Glucuronidación (UGT1A3 y UGT1A8)(No es sustrato de P-gP) | CYP3A4 (débil) | CYP2C9/19 (débil)P-gp (débil) | |
| Asociaciones contraindicadas o no recomendadas con etravirina | ||||
| Antirretrovirales:DTG: etravirina solo debe emplearse con dolutegravir si se asocian a ATV/r, DRV/r ó LPV/r.EFVIP no potenciadosNVPRPVTPV/r(Nota: en combinación con DRV/r 600/100 mg/12h ETR debe administrarse 2 veces al día: 200 mg/12h) | VHC:(Nota: boceprevir reduce un 29% la Cmin de ETR, significado clínico no establecido; ETR puede asociarse con telaprevir sin ajuste de dosis) | Otros:CarbamacepinaClaritromicina (para tratamiento de MAC, valorar azitromicina)Clopidogrel (teóricamente: ETR podría reducir la formación del metabolito activo de clopidogrel, significado clínico no establecido)Dexametasona (considerar alternativa a dexametasona, especialmente en uso crónico)DiazepamFenitoínaFenobarbitalHypericum (hierba de San Juan)ItraconazolKetoconazolPosaconazolRifampicina | ||
| Rilpivirina | CYP3A4(No es sustrato de P-gP) | — | P-gp (in vitro; aunque no afectó la farmacocinética de digoxina, no se puede descartar acumulación de otros sustratos más sensibles de la P-gp como dabigaran: precaución)MATE-2K (in vitro; significado clínico incierto) | |
| Asociaciones contraindicadas o no recomendadas con rilpivirinaEn general, fármacos que puedan aumentar el metabolismo de RPV (inductores del CYP3A4) o alterar el pH gástrico (inhibidores de la bomba de protones contraindicados) (Nota: puede darse con antihistamínicos H2 tipo ranitidina: 12h antes o 4h después de rilpivirina; con antiácidos: 2h antes o 4h después de RPV)Nota: en voluntarios dosis mayores a las terapéuticas de RPV (75mg una vez al día y 300mg una vez al día) prolongaron el intervalo QTc en el ECG. A la dosis de 25 mg/24h no se produce efecto clínicamente relevante sobre el intervalo QT. Usar con precaución conjuntamente con medicamentos que tienen un riesgo conocido de torsade de pointes. | ||||
| Antirretrovirales:EfavirenzEtravirinaNevirapina | VHC:(Nota: puede asociarse con boceprevir y telaprevir. Dado que ambos aumentan los niveles de rilpivirina, precaución en pacientes con riesgo ↑QT) | Otros:CarbamacepinaClaritromicina (no hay datos: ↑RPV, ambos ↑QT, considerar azitromicina)Dexametasona vía sistémica (excepto dosis única)Eritromicina (no hay datos: ↑RPV, ambos ↑QT, considerar azitromicina)EsomeprazolFenitoínaFenobarbitalHypericum (hierba de San Juan)LansoprazolOmeprazolOxcarbacepinaPantoprazolRabeprazolRifabutina (Nota: puede asociarse RPV con rifabutina: ↑RPV a 50 mg/día)Rifampicina (Nota: puede asociarse RPV con rifabutina: ↑RPV a 50 mg/día) | ||
| IP | Atazanavir | CYP3A4GlucuronidaciónP-gp | CYP2C8 (con ATV/r). Una interacción clínicamente importante entre ATV/r y los sustratos del CYP2C8 es poco probableb | CYP3A4CYP2C8 (débil)bUGT 1A1b y UGT 1A3P-gp (moderado)c |
| Darunavir/r | Véase ritonavir | |||
| Fosamprenavir/r | Véase ritonavir | |||
| Indinavir/r | Véase ritonavir | |||
| Lopinavir/r | Véase ritonavir | |||
| Ritonavir | CYP3A4>CYP2D6P-gp | CYP1A2, 2B62C19 (potente) y en menor medida 2C92C8(in vivo), glucuronidación (UGT1A4)Autoinduce su propio metabolismo | CYP3A4 (potente) y 2D6 (potente)P-gp (potente inicialmente; puede reducirse en el transcurso del tiempo)c | |
| Saquinavir | Véase ritonavir | |||
| Tipranavir/r(en estado de equilibrio)d | CYP3A4P-gp | 1A2 (moderado)2C9 (leve)CYP2B6glucuronidaciónP-gp (efecto inhibidor significativo tras primera dosis y efecto inductor mínimo tras uso continuado) | CYP3A4 intestinal (potente)CYP3A4 hepático (moderado)2D6 (potente)CYP2C8e | |
| IP/r | Asociaciones contraindicadas o no recomendadas con IP potenciados con ritonavirCon ATV/r: fármacos que puedan alterar el pH gástrico: inhibidores de la bomba de protones (IBP) contraindicados. Si IBP inevitable, ATV/r 400/100 mg/24h y estrecha monitorización. La dosis diaria del IBP no debe exceder 20mg de omeprazol o su equivalente y debe administrarse 12h antes de ATV/r.) (Nota: en pacientes que no estén tomando tenofovir pueden administrarse antihistamínicos H2 sin superar una dosis equivalente a 20mg de famotidina 2 veces al día. Si fuese necesaria una dosis mayor, equivalente a 40mg de famotidina 2 veces al día como máximo, considerar ATV/r 400/100 mg/24h. En pacientes que estén tomando tenofovir, si se administra una antihistamínicos H2 dosificar ATV/r 400/100 mg/24h y no superar 40 mg/12h de famotidina o equivalente. Es preferible administrar ATV/r 10h después o 2h antes de los antihistamínicos H2. Los antiácidos deben administrarse 1h antes o 2h después de atazanavir)Evitar también fármacos de estrecho margen terapéutico que sean metabolizados mayoritariamente mediante CYP3A4 o CYP2D6, que pueden acumularse y presentar toxicidad importanteCon ATV/r, LPV/r y SQV/r: evitar fármacos que puedan prolongar el intervalo QT | |||
| Antirretrovirales:ATV/r + NVPATV/r + EFV(considerar ATV/r 400/200mg c/24h en naïve; evitar en pretratados)DRV/r + EFV: emplear DRV/r 600/100 mg/12h)DRV/r 600/100 mg/12h + ETR: emplear ETR 2 veces al día: 200 mg/12hLPV/r + EFV o NVP: considerar LVP/r 500/125mg/12h; 5 comp. de 100/25mg por toma | VHC:Nota: solo está permitido el uso de ATV/r con telaprevir, monitorizando ↑bilirrubina. Boceprevir + ATV/r: esta coadministración podría valorarse caso a caso cuando se considere necesario, en pacientes con cargas virales del VIH suprimidas y con cepas virales del VIH sin sospecha de resistencia al tratamiento frente al VIH (se alcanzan niveles similares al uso de ATV no potenciado)El resto de IP/r no deben asociarse a boceprevir o telaprevir | Otros:AlfuzosinaAmiodaronaAnfetamina, derivadosAnticonceptivos orales (Nota: con ATV/r puede emplearse un anticonceptivo oral que contenga mínimo 30μg deetinilestradiol)AvanafiloBudesonida (incluso inhalada)CloracepatoClozapinaColchicina (si insuficiencia renal o hepática) (Nota: si función renal o hepática normales, usar dosis reducidas de colchicina)(Nota: dexametasona en uso prolongado puede reducir las concentraciones de los IP; precaución)DextropropoxifenoDiazepamDerivados de ergotaminaDisulfiram (no asociar a soluciones de Norvir® y Kaletra®)EncainidaEstazolamÉxtasisFenitoína y fenobarbital (evitar o monitorizar estrechamente niveles de antirretroviral y antiepiléptico)FlecainidaFluracepamFusídico, ácidoFluticasona (valorar beclometasona)HalofantrinaHypericum (hierba de San Juan)Irinotecan: con ATV/rLidocaína sistémicaLovastatinaLumefantrinaMeperidina o petidinaMetanfetaminaMetronidazol con las soluciones de Norvir® y Kaletra® (posible reacción de tipo disulfiram)Midazolam oralPimozidaPiroxicamPropafenonaQuetiapinaQuinidinaRifampicina (valorar rifabutina 150 mg/día)RivaroxabanSalmeterolSertindolSildenafilo (hipertensión pulmonar)SimvastatinaTerfenadinaTriazolamVardenafiloVoriconazol | ||
| ATV no potenciado | Asociaciones contraindicadas o no recomendadas con atazanavir no potenciadoEn general, fármacos que puedan aumentar el metabolismo de atazanavir (inductores del CYP3A4) o alterar el pH gástrico (inhibidores de la bomba de protones [IBP] contraindicados) (Nota: puede darse con antihistamínicos H2 tipo ranitidina: 10h antes o 2h después de atazanavir; los antiácidos deben administrarse 1h antes o 2h después de atazanavir)Evitar fármacos que puedan prolongar el intervalo QT | |||
| Antirretrovirales:Didanosina en comp. tamponados: administrar ATV 2h antes ó 1h después de didanosinaEfavirenz (considerar ATV/r 400/100mg c/24h en naïve; evitar en pretratados)IndinavirNevirapinaTenofovir (emplear ATV/r) | VHC:No asociar atazanavir no potenciado con boceprevir o telaprevir (ausencia de datos) | Otros:AlfuzosinaBosentán (emplear ATV/r y ajustar dosis de bosentán)Buprenorfina (emplear ATV/r)CarbamacepinaClaritromicina: para MAI reducir 50% dosis de claritromicina; otras indicaciones: valorar azitromicinaColchicina (si insuficiencia renal o hepática) (Nota: si función renal o hepática normales, usar dosis reducidas de colchicina)Derivados de ergotaminaEsomeprazolFenitoínaFenobarbital(Nota: fluticasona: precaución especialmente en uso a largo plazo)Hypericum (hierba de San Juan)IrinotecanLansoprazolLovastatinaMidazolam oralOmeprazolOxcarbacepinaPantoprazolPimozidaRabeprazolRifampicinaSalmeterolSertindolSildenafilo (hipertensión pulmonar; valorar tadalafilo ajustando dosis)SimvastatinaTriazolam | ||
| Inhibidores de la fusión | Enfuvirtida | Otros medicamentos queAl ser un péptido se elimina mediante catabolismo proteico. No es sustrato ni influye en la actividad del citocromo P-450Ausencia de interacciones clínicamente importantes | ||
| Antagonistas CCR5 | Maraviroc | CYP3A4P-gp | Otros medicamentos que actúan sobre el CYP3A4 pueden modificar las concentraciones de maraviroc. Sin embargo, es poco probable que maraviroc modifique las concentraciones de otros fármacosMaravirocin vitroinhibe la P-gp. Maraviroc no afectó la farmacocinética de digoxina; sin embargo, no se puede descartar una acumulación de otros sustratos más sensibles de la P-gp como dabigatrán | |
| Nota: Solo se incluyen las alternativas a estos casos; no se incluyen otras asociaciones que requieren ajuste de dosis o mayor seguimiento: véanse otras fuentes de información. No se incluye información de fármacos retirados del mercado como astemizol, bepridilo o cisaprida, ni de los que todavía no están comercializados, como rifapentina).Asociaciones contraindicadas o no recomendadas con maraviroc:En general, combinaciones de 2 inductores enzimáticos importantes (p.ej: rifampicina + efavirenz) | ||||
| Antirretrovirales:FPV/r | VHC:(Nota: puede asociarse con boceprevir y telaprevir ajustando maraviroc a 150 mg/12h) | Otros:Hypericum (hierba de San Juan) | ||
| Inhibidores de la integrasa del HIV | Raltegravir | UGT1A1(No es sustrato del citocromo P-450)P-gp no juega un papel importante | — | In vitro: no inhibe UGT1A1 ni UGT2B7; sin embargo, según un estudio clínico,in vivopuede existir cierta inhibición de la UGT1A1No inhibe P-gp |
| Asociaciones contraindicadas o no recomendadas con raltegravirNo se recomienda su administración simultánea con antiácidos que contienen aluminio y/o magnesio: espaciar 4h. No se han descrito otras asociaciones contraindicadas o no recomendadas con raltegravir. Con rifampicina, la ficha técnica del producto recomienda valorar aumento de dosis de RAL a 800mg/12h; se está investigando la posibilidad de mantener la dosis estándar. No hay datos con otros inductores potentes del UGT1A1 como carbamazepina, fenobarbital o fenitoína.(Nota: raltegravir puede asociarse a telaprevir y boceprevir sin que se requiera ajuste de dosis) | ||||
| Dolutegravir | UGT1A1 (mayoritario)CYP3A4 (minoritario)In vitroes sustrato de UGT1A3, UGT1A9 y de los transportadores BCRP y P-gp | Transportadores OCT2 y MATE.(La inhibición de los transportadores OAT1 y 3 probablemente carezca de importancia clínica in vivo)bP-gp: una interacción parece poco probableNo inhibe UGT1A1 ni 2B7 | ||
| Asociaciones contraindicadas o no recomendadas con dolutegravirEn general, fármacos que puedan aumentar su metabolismo (inductores del CYP3A4) o que contengan cationes como los antiácidos y sean administrados simultáneamente (debe administrarse dolutegravir 2h antes o 6h después de los cationes) | ||||
| Antirretrovirales:Etravirina (si no va asociada a IPs)Nota: con inductores como efavirenz, nevirapina o tipranavir/ritonavir dosificar dolutegravir 50 mg/12h o considerar alternativas. Esta pauta solo se recomienda en ausencia de resistencia a inhibidores de la integrasaFPV/r: evitar en presencia de resistencia a inhibidores de la integrasa. En ausencia de resistencia no se requiere ajuste de dosis | VHC:(Nota: dolutegravir puede asociarse a telaprevir y boceprevir sin que se requiera ajuste de dosis) | Otros:CarbamacepinaFenitoínaFenobarbitalHypericum (hierba de San Juan)OxcarbazepinaMedicamentos que contengan cationes: antiácidos, laxantes, sucralfato, hierro, calcio, administrados simultáneamente (debe administrarse dolutegravir 2h antes o 6h después)Nota: con rifampicina, dosificar dolutegravir 50 mg/12h o considerar alternativas. Esta pauta solo se recomienda en ausencia de resistencia a inhibidores de la integrasa | ||
| Elvitegravir(ver también cobicistat, que se emplea como potenciador farmacocinético) | CYP3A4 (mayoritario)UGT1A1/1A3 (minoritarios) | Inductor de escasa potencia sobre CYP2C9 y UGTInductor débil de CYP3A4 | Inhibidor débil de P-gp | |
| Potenciador fármaco-cinético | Cobicistat | CYP3A4 (mayoritario)CYP2D6 (minoritario)(no es sustrato de UGT) | CYP3A4 (potente)CYP2D6 (débil)P-gp (potencia similar a ritonavir)BCRP en intestinoOATP1B1 y OATP1B3 en hígado | |
| Asociaciones contraindicadas o no recomendadas con elvitegravir/cobicistat:En general, evitar fármacos que puedan reducir su eficacia (inductores del CYP3A4) o los antiácidos (espaciando 4h el antiácido se evita esta interacción)Evitar también fármacos de estrecho margen terapéutico que sean metabolizados mayoritariamente mediante el CYP3A4, que pueden acumularse y presentar toxicidad importante | ||||
| Antirretrovirales:Stribild® es una combinación de ARV, por lo que no debe asociarse a otros ARV | VHC:(Nota: elvitegravir puede asociarse a telaprevir sin que se requiera ajuste de dosis; no hay datos con boceprevir) | Otros:AlfuzosinaAnticonceptivos orales: utilizar un anticonceptivo hormonal que contenga mínimo 30μg de etinilestradiol y que contenga norgestimato como progestágeno, o usar un método alternativo fiable. No hay datos con otros progestágenos: evitar.Antiácidos de Al/Mg simultáneamente (espaciar 4h)Atorvastatina (evitar o valorar beneficio/riesgo y emplear la mínima dosis posible)BosentánCarbamacepinaColchicina (si insuficiencia renal o hepática) Nota: en pacientes con función renal y hepática normal puede requerir ajuste de dosis)Derivados de la ergotaminaFenobarbitalFenitoína(Nota: Fluticasona precaución y estrecha monitorización; preferiblemente beclometasona)Hypericum (hierba de San Juan)LovastatinaMidazolam por vía oralOxcarbazepinaPimozidaRifabutinaRifampicinaSalmeterolSertindolSildenafilo (en hipertensión pulmonar)SimvastatinaSuplementos vitamínicos simultáneamente (espaciar 4h)TriazolamVoriconazol (evitar o valorar riesgo/beneficio) | ||
Nota: Solo se incluyen las alternativas a estos casos; no se incluyen otras asociaciones que requieren ajuste de dosis o mayor seguimiento: véanse otras fuentes de información. No se incluye información de fármacos retirados del mercado como astemizol, bepridilo o cisaprida, ni de los que todavía no están comercializados, como rifapentina).
Potencia inhibidora inversamente proporcional a la IC50: LPV/r 10,3 mcM > NFV 19,9 mcM > ritonavir 39,6 mcM > ATV 67,8 mcM > SQV 100 mcM > APV/IDV/DRV > 100 mcM.
Los efectos de TPV/r sobre el CYP3A4 y la P-gp son complejos y diferentes tras la primera dosis o en estado de equilibrio estacionario (dosis múltiples). Tras la primera dosis inhibe de forma leve el CYP1A2 y el CYP2C9, inhibe de forma moderada el CYP3A4/5 hepático, el CYP2D6 y la P-gp intestinal e inhibe de forma potente el CYP3A4 intestinal. En estado de equilibrio inhibe de forma moderada del CYP3A4/5 hepático, inhibe de forma potente el CYP2D6 y el CYP3A4/5 intestinal, induce el CYP1A2 y el CYP2C9 y tiene efecto mínimo inductor sobre la P-gp. El efecto neto de TPV/r sobre las concentraciones plasmáticas de los fármacos que son sustratos del CYP3A4 y de la glucoproteína-P dependerá de la afinidad por los mismos y es difícil de establecer.
Existen diversas páginas web que permiten consultar posibles interacciones182-184. Debido a que la información científica relacionada con los FAR se renueva constantemente, se recomienda consultar también la ficha técnica y la información actualizada por las compañías farmacéuticas y las autoridades sanitarias185,186.
Algunas interacciones farmacodinámicas (modificación del efecto a nivel de receptor sin cambios en las concentraciones) tienen interés clínico. Dan lugar a adición, sinergia o antagonismo, ya sea reduciendo o aumentando la eficacia o la toxicidad. Algunos ejemplos son: el incremento del riesgo de toxicidad mitocondrial al asociar ribavirina con ddI, de toxicidad renal al asociar TDF a otros fármacos nefrotóxicos, o de anemia al asociar ZDV con los IP que actúan contra el VHC, boceprevir y telaprevir, más interferón y ribavirina.
Se han descrito interacciones importantes entre los IP con acción frente al VHC y los FAR, que pueden consultarse en una página web específica187, así como en las recomendaciones elaboradas por la Agencia Española del Medicamento y Productos Sanitarios188.
Recomendaciones- –
Se deben reseñar en la historia clínica todos los medicamentos, productos naturales y medicinas alternativas que toma el paciente para evaluar posibles interacciones entre ellos (A-III).
- –
Se deben tener en cuenta las contraindicaciones y realizar ajustes de dosis cuando sea necesario (A-I).
- –
Se debe considerar la monitorización de los niveles plasmáticos cuando se administren 2 o más fármacos con posibles interacciones farmacocinéticas relevantes, para evitar toxicidad o ineficacia terapéutica (A-II).
La infección aguda por VIH-1 se caracteriza en más de la mitad de los casos por un síndrome febril agudo autolimitado, similar a la gripe o a la mononucleosis infecciosa189,190. Las pruebas de ELISA de cuarta generación, que incluyen la determinación simultánea de anticuerpos y del antígeno p24 del VIH-1, acortan el periodo ventana y permiten el diagnóstico en el 80-90% de casos. La infección aguda (primeros 30 días) no debe confundirse con la infección reciente, que hace referencia a pacientes diagnosticados en los primeros 6 meses (180 días) de la infección.
El inicio del TAR en la infección aguda tiene ventajas importantes189,190, como acortar la duración y la gravedad de los síntomas, suprimir la replicación viral, reducir la diversidad viral y el reservorio (ADN proviral), reducir el riesgo de transmisión del VIH-1, normalizar la cifra de linfocitos CD4+, reducir la activación inmunológica y preservar o restaurar la inmunidad específica frente al VIH-1, lo que, en teoría, podría permitir el control inmunológico de la replicación viral sin TAR. Por el contrario, las desventajas del TAR son: la exposición a los FAR sin un beneficio clínico claramente demostrado, el riesgo de efectos secundarios, su duración indefinida, el desarrollo de resistencias si el cumplimiento no es adecuado, y el tratamiento innecesario de los individuos controladores de elite. Sin embargo, en la actualidad disponemos de regímenes fáciles de administrar (un comprimido al día), potentes y seguros, lo que facilita el TAR de estos pacientes.
Las estrategias terapéuticas para evitar un TAR indefinido, que se han realizado en los últimos años, han fracasado189,190: a)administrar el TAR durante un periodo limitado de tiempo (ensayos clínicos SPARTAC, PRIMO-SHM y ACTG A5217); b)administrarlo de forma intermitente, a fin de potenciar la respuesta VIH-1-específica y controlar la replicación viral sin FAR; c)combinar el TAR con inmunosupresores (hidroxiurea, ciclosporinaA, ácido micofenólico) o citoquinas (IL-2, interferón), y d)asociar vacunas terapéuticas (ensayos clínicos QUEST, ACTG A5187) al TAR. En la mayoría de los pacientes ninguna de estas estrategias ha conseguido que el sistema inmune controle la replicación viral de forma sostenida sin TAR, y el beneficio inmunológico y virológico alcanzado no ha durado más de 2años191. Solo se ha observado control virológico en el 4-8% de los pacientes192 que en general iniciaron el TAR precozmente (<90días), pero paradójicamente las características virológicas, inmunológicas y genéticas de estos «controladores post-TAR»193 son completamente diferentes a los controladores de elite. Por tanto, en la práctica clínica, si se inicia el TAR durante la infección aguda, este se debe mantener de forma indefinida. Las pautas de TAR deben ser las mismas que en la infección crónica, y la adición de más FAR (4 o 5) no está recomendada, ya que no se ha acompañado de ningún beneficio adicional194.
El TAR en la infección aguda/reciente debe iniciarse lo antes posible y preferiblemente en los primeros 4 meses de la infección por el VIH, ya que es cuando se puede conseguir el máximo beneficio inmunológico (alcanzar más de 900 linfocitos CD4/μl)195.
Finalmente, siempre debe valorarse la inclusión de estos pacientes en protocolos de investigación o ensayos clínicos que busquen la erradicación o la cura funcional del VIH-1.
Recomendaciones- –
El TAR debe recomendarse en todos los pacientes con infección aguda por el VIH, independientemente de los síntomas, su gravedad y su duración (A-II), y debe comenzarse tan pronto como sea posible para obtener el máximo beneficio.
- –
En general, el TAR debe ofrecerse a todos los pacientes con infección reciente (B-II). En los pacientes con una cifra menor de 500 linfocitos CD4/μl debe comenzarse en los primeros 4 meses para obtener el máximo beneficio.
- –
Se debe recomendar iniciar el TAR en todos los casos en los que exista un alto riesgo de transmisión del VIH-1 (A-II).
- –
Se debe recomendar el inicio del TAR cuando la infección aguda por el VIH ocurre durante el embarazo (A-I).
- –
El inicio del TAR se hará con las mismas pautas preferentes recomendadas para la infección crónica por el VIH-1 (A-I) (tabla 3). Una pauta con 2 ITIAN (preferentemente TDF/FTC) y un INI podría reducir más rápidamente la CVP durante las primeras 4-8 semanas en comparación con los IP o ITINN (A-I), lo que podría facilitar la reducción de la transmisión del VIH. La combinación de RAL+2ITIAN (preferentemente TDF/FTC) podría tener la ventaja de alcanzar concentraciones más elevadas en las secreciones genitales (B-III).
- –
Se debe efectuar siempre una prueba de resistencia y un tropismo viral al diagnóstico de la infección aguda o reciente, se vaya a iniciar TAR o no (A-II).
- –
Si no se dispone del resultado del estudio de resistencia es preferible comenzar con una pauta basada en un IP/r hasta tener los resultados (A-II).
- –
Una vez iniciado el TAR, debe administrarse por tiempo indefinido (A-I).
El VIH-2 posee una organización genómica similar al VIH-1, aunque con diferencias estructurales que influyen de forma significativa en su patogenicidad y en su sensibilidad a los FAR196.
Diversas circunstancias provocan que la toma de decisiones acerca del TAR en los pacientes con infección por el VIH-2 tenga una mayor dificultad: a)la historia natural del VIH-2 difiere de la del VIH-1, y carecemos de las evidencias necesarias que permitan identificar el momento óptimo de inicio del TAR en estos pacientes; b)los algoritmos genotípicos utilizados para predecir resistencias a FAR en la infección por VIH-1 no son directamente aplicables al VIH-2; c)no disponemos de evidencias procedentes de ensayos clínicos que permitan identificar los regímenes de uso preferencial en el escenario de la infección por VIH-2; d)en el momento actual tampoco disponemos de una prueba comercial para medir la carga viral del VIH-2, ni de pruebas de resistencia fenotípicas o genotípicas validadas para uso clínico. Pese a estas limitaciones, parece razonable asumir que los principios generales del TAR en los pacientes con infección por el VIH-2 deben ser los mismos que para el VIH-1.
El VIH-2 presenta importantes diferencias respecto al VIH-1 en su perfil de sensibilidad a los FAR. El VIH-2 presenta resistencia intrínseca a los ITINN197. En contraposición, es sensible a los ITIAN, aunque la barrera genética frente a la resistencia de estos es más baja que frente al VIH-1198. Además, el VIH-2 presenta una sensibilidad variable frente a los IP, siendo LPV, SQV y DRV los más activos199. RAL tiene actividad in vitro frente al VIH-2200. El uso de MVC en el tratamiento de la infección por VIH-2 está limitado por la no disponibilidad de un test de tropismo y por la capacidad del VIH-2 para utilizar, además del CCR5 y el CXCR4, otros correceptores201. Por último, el VIH-2 presenta resistencia intrínseca a ENF202. En este escenario, hasta no disponer de nuevas evidencias, parece razonable recomendar, como régimen preferente para el TAR de inicio en pacientes infectados por el VIH-2 o con infección dual por VIH-1/2, la combinación de 2 ITIAN y un IP/r.
Recomendaciones- –
Los principios generales del TAR en pacientes con infección por el VIH-2 deben ser los mismos que para la infección por el VIH-1 (A-III).
- –
El régimen de TAR de inicio de uso preferente en estos pacientes es la combinación de 2 ITIAN+1IP/r (A-III).
- –
El uso de ITINN, MVC o ENF está contraindicado para el tratamiento de la infección por el VIH-2 (A-I).
El TAR en el embarazo se discute en un documento de consenso elaborado por el PNS en colaboración con GeSIDA y otras sociedades científicas203. Se recomienda su lectura y la de otra guía internacional sobre el tema204, así como otras aportaciones205-209, para cualquier duda al respecto. En este apartado nos limitamos a presentar las recomendaciones acerca del TAR en este contexto. El manejo obstétrico y del recién nacido está fuera de los objetivos y del alcance de esta guía.
Recomendaciones- –
Es imprescindible realizar una serología de VIH en toda mujer embarazada (A-I) y, si fuera negativa, repetirla en el tercer trimestre (A-II).
- –
El consejo preconcepcional debe formar parte de la asistencia a la mujer con infección por el VIH en edad reproductiva (A-II).
- –
El TAR está indicado en todas las gestantes, independientemente del número de linfocitos CD4+ y de la CVP que presenten con el objetivo de mantener una CVP indetectable (A-I).
- –
La elección de los FAR concretos se basará en el estudio de resistencia y en la seguridad de los mismos (véanse tablas 10–12). Si no hay resistencias, el TAR de elección es ZDV o TDF o ABC+3TC o FTC+LPV/r o ATV/r (A-I); en caso contrario, podrán recibir cualquiera de los FAR «recomendados» o «alternativos» tras una valoración individualizada (A-III). En la tabla 11 se recoge la actitud recomendada ante diferentes situaciones.
Tabla 10.Fármacos antirretrovirales y embarazo
Fármaco / Categoría FDA Toxicidad potencial enla gestación Comentarios ITIANZDV / CD4T / CDDI / BABC / C3TC / CFTC / BTDF / B MielotoxicidadToxicidad mitocondrialReacción de hipersensibilidadNefrotoxicidadAlteración del metabolismo P/Ca ZDV: amplia experiencia, su uso es seguro en el embarazoD4T + DDI: incremento del riesgo de muerte materna por acidosis láctica y fallo hepáticoABC: reacción de hipersensibilidad potencialmente grave en presencia de mutación HLA B5701TDF: si se opta por TDF, vigilar función renal ITINNEFV / DNVP / BETR / BRPV / B Alteraciones SNCHepatotoxicidad EFV: contraindicado por riesgo de anencefalia y defectos del tubo neural en primates y humanos en primer trimestre de la gestaciónNVP: amplia experiencia en el empleo durante la gestación; riesgo de hepatotoxicidad grave con CD4 > 250 células/μl, especialmente en coinfección por VHB y VHC. Este efecto adverso no es extrapolable en mujeres que ya estaban en TAR con NVP al inicio de la gestación y/o que han tomado NVP previamente IP/rRitonavir / BLPV / CSQV / BATV / BDRV / CFPV / CTPV / C HiperglucemiaRiesgo de prematuridad ATV: Riesgo de hiperbilirrubinemia neonatal, aunque parece poco relevante en estudios realizados en embarazadasSQV y LPV: bien tolerados en el embarazo Inhibidores de la fusiónENF / B Datos escasos en gestación Inhibidores integrasaRAL / CEVG / BDTG / B Datos escasos en gestaciónSin datos en gestaciónSin datos en gestación Inhibidores CCR5MVC / B Datos escasos en gestación Tabla 11.Manejo del tratamiento antirretroviral durante la gestación en distintas situaciones
Semana de inicio del TAR Fármacos Comentarios Gestante sin tratamiento y sin indicación de TAR A partir de las 12-14 semanasa IP/r + 2 ITIAN En caso de interrupción de TAR con IP + 2 ITIAN, se interrumpirán todos los fármacos a la vez Gestante sin tratamiento y con indicación de TARb Inicio lo más precoz posible IP/r + 2 ITIAN, o bienNVP + 2 ITIAN(esta última solo si CD4 < 250 células/μl) En caso de cambio de TAR compuesto por NVP + 2 ITIAN durante la gestación motivado por toxicidad o intolerancia, mantener los 2 ITIAN y añadir IP/r a los 7 diasc Gestante en TAR Mantener TARSi la gestante interrumpió el TAR, reiniciar a las 12-14 semanasa Sustituir fármacos teratogénicos (EFV)Evitar combinaciones con riesgo elevado de toxicidad (D4T + DDI) o menor eficacia (3 ITIAN)Si está con NVP, mantener aunque CD4 > 250 células/μl En pautas de TAR con LPV/r se debe administrar BIDSi ATV/r se acompaña de TDF o fármacos anti-H2, aumentar la dosis de ATV a 400mg en el 2.° y 3.er trimestre Gestante con indicación de TAR y antecedentes de abandono del TARb Inicio lo más precoz posible Pauta de TAR según estudios de resistencias y TAR previosSi coinfección por VHB, mantener 3TC o considerar TDF + FTC Utilizar como primera opción los antirretrovirales con mayor experiencia siempre que sea posible Gestante con primoinfección por VIHb Inicio en el momento del diagnóstico IP/r + 2 ITIN. Ajustar en cuanto se conozca el genotipo Si el diagnóstico es en el tercer trimestre, programar cesárea electiva Gestante con infección por VIH desconocida en el parto Realizar test rápido para VIH Si positivo, administrar ZDV i.v. + NVP oral en dosis única y proceder con cesárea electiva Consultar documento específico para ver tratamiento a administrar al RN y cómo seguir en la madre Mujer en TAR que desea quedar embarazada Evitar EFV Insistir en la adherencia y en mantener la CV indetectable previa al embarazo aEn mujeres con carga viral elevada o con indicación de TAR por salud materna, debe considerarse iniciar lo más precozmente posible, siempre que se comience con fármacos seguros y que no haya una hiperémesis gravídica incontrolable.
bCuando en esta situación la presentación es próxima al parto, se puede valorar también la adición de raltegravir, dado su rápido paso placentario, a dosis de 400mg/12h, aunque la experiencia con este fármaco es escasa; y/o de NVP oral al menos 2h antes del parto asociada a AZT i.v. (véase documento específico para ampliar la información).
Tabla 12.Recomendaciones de uso de fármacos antirretrovirales en el embarazo
Recomendados Alternativosa No recomendados (salvo que no haya otra alternativa) Contraindicados ITIAN ZidovudinaLamivudinaAbacavirbTenofovircEmtricitabina DidanosinaEstavudina Didanosina + estavudinad ITINN Nevirapinae, si CD4 < 250 células/μl Efavirenz en segundo y tercer trimestresf Efavirenz en primer trimestref IP/r Lopinavir/r, 400/100 BID Darunavir/r, 600/100 BIDAtazanavir/rg 300/100h QDSaquinavir/r1.000/100 BID Fosamprenavir/r Inhibidores de la fusión Inhibidores de la integrasa Raltegravir (RAL) 400 c/12 h aUsar cuando no puedan utilizarse los fármacos de 1.a elección. No hay datos para etravirina, rilpivirina, elvitegravir, dolutegravir, tipranavir ni enfuvirtida.
cRiesgo potencial de alteraciones renales y óseas y alteraciones del metabolismo calcio-fósforo tanto en animales como en pacientes VIH. Debe considerarse especialmente para el tratamiento en pacientes con confección por VHB. Además, datos recientemente publicados sugieren que es un fármaco eficaz y probablemente seguro.
eMayor riesgo de hepatotoxicidad en gestantes coinfectadas por VHC, VHB o con linfocitos CD4 > 250 células/μl.
fCategoría D, potencialmente teratógeno, aunque datos observacionales sugieren la posibilidad de mantenerlo, especialmente si el diagnóstico de embarazo se hace a partir de la 5.a-6.a semana, y sobre todo la posibilidad de utilizarlo electivamente a partir de la 8.a-9.a semana.
gHiperbilirrubinemia, riesgo potencial de kernicterus. En adultos se ha descrito un aumento de la bilirrubina no conjugada con el uso de este fármaco. Sin embargo, en una pequeña serie de 40 gestantes tratadas con este fármaco no se ha descrito una mayor incidencia de hiperbilirrubinemia en los neonatos con respecto a la población general.
- –
El tratamiento intraparto con ZDV por vía intravenosa está indicado en mujeres con CVP mayor a 1.000 copias o desconocida en el momento del parto, con independencia del TAR que hubiese llevado previamente la paciente (A-I).
- –
La cesárea electiva está indicada, en la semana 38, en mujeres cuya CVP previa al parto es mayor de 1.000 copias/ml (A-II).
- –
Las madres deben abstenerse de forma absoluta de lactar a sus hijos y deberán alimentarlos con una fórmula adaptada (A-I).
En la actualidad se recomienda iniciar el TAR en fases precoces de la infección por el VIH. A pesar de ello, una proporción considerable de pacientes acude por primera vez en fases avanzadas, con inmunodepresión profunda y, en ocasiones, presentando enfermedades oportunistas, la mayoría infecciosas (IO).
El momento idóneo para iniciar el TAR en un paciente con una IO es motivo de controversia210. Las posibles ventajas de un inicio temprano incluyen una recuperación inmune más rápida, una mayor resolución de la IO, prevenir la aparición de otras y reducir el riesgo de mortalidad. Entre los inconvenientes del inicio precoz del TAR están el número elevado de comprimidos a tomar para el tratamiento concomitante de la IO y del VIH, posibles interacciones y toxicidades, y el síndrome inflamatorio de reconstitución inmune (SIRI)210. La IO en la que se ha estudiado mejor el momento más idóneo para iniciar el TAR es la tuberculosis (TB), que se trata en otro apartado. Existen pocos estudios aleatorizados y de cohortes que hayan evaluado este tema en otras IO. El ensayo clínico aleatorizado ACTG A5164 demostró una reducción en la progresión a sida/muerte (OR: 0,51; IC95%: 0,27 a 0,94) en pacientes que iniciaban TAR dentro de una mediana de 12días tras el inicio del tratamiento de la IO frente a los que lo demoraban hasta una mediana de 45 días211. No se observaron diferencias significativas en efectos adversos, incidencia de SIRI ni su aparición según se usasen o no esteroides para la IO. Más del 60% de los pacientes tenían neumonía por Pneumocystis jiroveci, pero dado el pequeño número y variedad de otras IO es difícil extraer conclusiones para cada una de ellas por separado. En todo caso, parecía haber una tendencia favorable al inicio precoz en las infecciones fúngicas, incluyendo la meningitis criptocócica (MC).
Un estudio prospectivo de cohorte, realizado por el grupo PISCIS, halló también una mayor progresión a sida/muerte en los pacientes que iniciaron el TAR entre 30-270 días tras el comienzo del tratamiento de la IO, frente a los que lo hicieron antes de 30 días, tanto en IO en general como en la P.jiroveci (p=0,002; RR: 1,83; IC95%: 1,25 a 2,68)212.
Una de las IO más complicadas de tratar por su elevada morbimortalidad es la MC, sobre todo en los países empobrecidos. La ausencia de un tratamiento estandarizado para el aumento de la presión intracraneal, la falta de 5-fluorocitosina y la dificultad para acceder al TAR en esos países contribuyen seguramente a su mayor mortalidad.
Algunos estudios han intentado evaluar la importancia del momento de iniciar el TAR en pacientes con MC. En Zimbabwe, Makadzange et al.213 observaron una peor supervivencia en los pacientes que iniciaron el TAR (d4T/3TC/NVP) antes de 72h del comienzo del tratamiento de la MC respecto a los que lo iniciaron después de 10 semanas (RR: 2,85; IC95%: 1,1 a 7,23). Sin embargo, en dicha mala evolución pudo influir la ausencia de un protocolo de manejo de la hipertensión craneal, el uso de fluconazol en vez de anfotericinaB y una carga antigénica más elevada en el momento del inicio del TAR en el grupo de inicio inmediato.
En un reciente estudio aleatorizado en Botswana, Bisson et al.214 evaluaron a 27 pacientes con MC que iniciaron TAR con una mediana de 7 días frente a 32 tras la aleatorización; todos los pacientes llevaban tratamiento con anfotericinaB. No encontraron diferencias en el tiempo hasta la negativización del cultivo del criptococo en el LCR, aunque sí una tendencia a menor mortalidad a pesar de una mayor frecuencia de SIRI en los pacientes con inicio precoz del TAR. No obstante, este estudio tiene la limitación de su pequeño tamaño muestral, menor al número previsto en el diseño por problemas de selección.
Finalmente, en el estudio Cryptococcal Optimal ART Timing (COAT), organizado por la Universidad de Minnesota y realizado en África, se detuvo la inclusión de pacientes y el estudio por una mayor mortalidad si el TAR se iniciaba precozmente215. Los pacientes (n=177 y con medianas de linfocitos CD4+ de 19 y 28 células/μl, respectivamente) fueron aleatorizados a inicio precoz o tardío del TAR (1-2 semanas o 5 semanas después del diagnóstico) y recibieron anfotericinaB (0,7-1,0mg/kg/día) y fluconazol (800mg/día) seguido de una consolidación con fluconazol. La mortalidad a las 26 semanas fue significativamente mayor en los que iniciaron tratamiento de forma precoz (45% vs. 30%; RR: 1,73; IC95%: 1,06 a 2,82; p=0,03). La mortalidad fue particularmente elevada en pacientes con menos de 5 células/μl en el LCR. La presencia de SIRI diagnosticado no difirió significativamente entre ambos grupos (20% vs. 13%; p=0,32). Los autores concluyeron que, en pacientes con MC, diferir el TAR 5semanas tras el diagnóstico se asociaba de forma significativa con una mayor supervivencia, comparado con aquellos en los que se inició el TAR 1-2semanas tras el diagnóstico, especialmente en pacientes con poca celularidad en el LCR215.
Por tanto, a pesar de la limitación de los datos, en las IO en general habría una menor progresión de la enfermedad en aquellos pacientes que inician el TAR de forma precoz (antes de los 30días), aunque no está claro si, además de para P.jiroveci, esto es cierto para todas las IO. Concretamente, en la MC (probablemente en relación con la gravedad del SIRI en el SNC, no siempre clínicamente reconocido, como también ocurre en la meningitis tuberculosa) sería mejor esperar varias semanas tras el diagnóstico y tratamiento de esta IO para iniciar el TAR215.
Recomendaciones- –
En pacientes con IO se debe iniciar el TAR dentro de los primeros 15-30 días desde el inicio del tratamiento de la infección (A-II).
- –
En el caso de la meningitis criptocócica, es prudente esperar varias semanas (5 según el estudio más amplio publicado hasta ahora, especialmente en pacientes con menos de 5 células/μl en el LCR) antes de iniciar el TAR (A-I). La negatividad del cultivo del LCR es un dato que apoya el inicio del TAR (A-II), pero, en general, debe considerarse cuando se observe un descenso de la carga antigénica (B-III). Además, se debe seguir un control estrecho de la hipertensión intracraneal (A-I).
El tratamiento de la TB en adultos infectados por el VIH ha sido objeto de un documento de consenso reciente de GESIDA/Secretaría del PNS216. Se recomienda su lectura para cualquier duda relacionada con el tratamiento de la TB o de la infección por VIH en pacientes que padecen las 2 enfermedades. En este apartado nos limitamos a presentar de modo resumido los aspectos más relevantes del TAR en este contexto.
Momento de inicio óptimo del TAR en pacientes infectados por VIH con TB. Esta importante cuestión se ha visto resuelta gracias a 3 ensayos clínicos realizados en diferentes partes del mundo, cuyos resultados fueron, con pequeñas diferencias, congruentes entre sí217-219.
Recomendaciones- –
Se recomienda iniciar siempre el TAR durante el tratamiento de la TB, independientemente del recuento de células CD4+, ya que se disminuye el riesgo de muerte (A-I).
- –
El momento óptimo de iniciar el TAR depende del recuento de células CD4+. Si el recuento es menor de 50 células/μl, debe iniciarse lo antes posible, tras comprobar la tolerabilidad al tratamiento antituberculoso y no más tarde de las 2 primeras semanas (A-I). Si el recuento de CD4+ es mayor de 50 células/μl, puede retrasarse el inicio del TAR hasta finalizar la fase intensiva del tratamiento antituberculoso (8 semanas) (A-I).
- –
Con las recomendaciones previas se disminuye el riesgo de efectos adversos y el desarrollo de SIRI, sin comprometer la supervivencia (A-I).
Pautas de TAR. La principal dificultad en el tratamiento adecuado simultáneo de la TB y la infección por VIH radica en las posibles interacciones medicamentosas. Estas son especialmente relevantes en el caso de las rifamicinas, por ser potentes inductores del sistema enzimático CYP3A4, implicado en el metabolismo de la mayoría de las familias de FAR.
Como principio general debe intentarse, siempre que sea posible, incluir rifampicina en el tratamiento de la TB y usar un régimen de TAR con fármacos sin problemas de interacciones. Para el paciente que inicia TAR, la elección del régimen debe seguir las mismas normas que para la población sin TB y debe incluir, por tanto, 2 ITIAN y un tercer fármaco. Para pacientes que han recibido previamente TAR y han desarrollado resistencia o intolerancia, el régimen debe conformarse de acuerdo con los principios para la elección de FAR en pacientes en tratamiento para TB que se resumen a continuación. Las recomendaciones pueden sintetizarse en los siguientes puntos220-224:
Recomendaciones- –
Elección de los ITIAN. No existe interacción significativa entre los fármacos antituberculosos y los ITIAN, ni hay evidencia de la potenciación de la toxicidad entre ellos. Por tanto, ABC, TDF, 3TC o FTC pueden ser utilizados en estos pacientes sin riesgos añadidos (A-I).
- –
Elección del tercer fármaco. La mayor experiencia y los mejores resultados se han obtenido con EFV, por lo que este constituye el FAR de elección (A-I). La dosis de EFV es la estándar en todos los pacientes (600mg/día), independientemente del peso, sin necesidad de aumentarla a 800mg/día (A-I).
- –
Alternativas terapéuticas como terceros fármacos. Existe experiencia o evidencia suficiente para recomendar como alternativas pautas que incluyan NVP a dosis habituales (A-II), RAL a dosis de 800mg/12h (A-II), aunque la dosis de 400mg/12h ha demostrado eficacia, y MVC a dosis de 600mg/12h (A-III).
- –
Fármacos que no pueden utilizarse. No se pueden coadministrar con rifampicina los otros ITINN (RPV y ETR), ningún IP (potenciado o no con ritonavir) ni EVG. En el caso excepcional de que un IP fuese la única opción de TAR, debe sustituirse rifampicina por rifabutina y realizar el ajuste correspondiente en las dosis de los fármacos (A-I).
El SIRI es una complicación frecuente del TAR en pacientes con TB, especialmente en pacientes con recuentos de células CD4+ muy bajos y cuando el TAR se inicia muy precozmente en relación con el inicio del tratamiento antituberculoso225.
Uso de fármacos antirretrovirales en pacientes con insuficiencia renal (tabla 13)Para una visión completa del diagnóstico, la prevención y el tratamiento de las alteraciones renales en pacientes con infección por VIH se recomienda consultar el documento de consenso ad hoc elaborado por GESIDA, la SEN y la SEQC226.
Ajuste de dosis de los antirretrovirales en insuficiencia renal y en hemodiálisis
| Familia | Fármacos | Dosis habituales | Dosis en insuficiencia renal | Dosis en hemodiálisis/diálisis peritoneal |
|---|---|---|---|---|
| Inhibidores de la transcriptasa análogos de nucleósidos/nucleótidos | Abacavir (ABC) | 300 mg/12h | No requiere ajuste de dosisNo administrar Combivir® y Trizivir® en pacientes con Cl < 50 ml/min (por separado, ajustar dosis adecuadamente) | Dosis habitualHD: administrar independientemente de la sesión de HD, ya que se elimina mínimamente |
| Didanosina, cápsulas entéricas (ddI) | Peso ≥60kg 400 mg/24h | ≥60 kgCl ≥60: 400mg c/24hCl 30-59: 200mg c/24hCl 10-29: 150mg c/24hCl < 10: emplear Videx® polvo para solución pediátrica 100mg c/24h | HD/CAPD: 100mg c/24h; los días de HD administrar post-HD/CAPD (no requiere suplemento) | |
| Peso < 60kg 250 mg/24h | < 60 kgCl ≥60: 250mg c/24hCl 30-59: 150mg c/24hCl 10-29: 100mg c/24hCl < 10: emplear Videx® polvo para solución pediátrica 75 mg/24h | HD/CAPD: emplear Videx® polvo para solución pediátrica 75 mg/24h | ||
| Estavudina (d4T) | ≥ 60 kg: 40 mg/12h | ≥ 60 kgCl ≥ 50: 40mg c/12hCl 26-49: 20mg c/12hCl ≤ 25: 20mg c/24h | HD: 20mg c/24h; los días de HD administrar post-HD | |
| < 60 kg: 30 mg/12h | < 60 kgCl ≥ 50: 30mg c/12hCl 26-49: 15mg c/12hCl ≤ 25: 15mg c/24h | HD: 15mg c/24h; los días de HD administrar post-HD | ||
| Lamivudina (3TC) | 150 mg/12h o 300 mg/24h | Cl ≥ 50: 150mg c/12h o 300mg c/24hCl 30-49: 150mg c/24h (primera dosis de 150 mg)Cl 15-29: 100mg c/24h (primera dosis de 150 mg)Cl 5-14: 50mg c/24h (primera dosis de 150 mg)Cl < 5: 25mg c/24h (primera dosis de 50 mg)No administrar Combivir® y Trizivir® si Cl <50 ml/min (administrar los componentes por separado, ajustando dosis adecuadamente) | HD: 25mg c/24h (primera dosis de 50 mg). Los días de la HD, administrar post-HD | |
| Emtricitabina (FTC) | 200mg c/24h (cápsulas)240mg c/24h (solución oral) | En cápsulas:Cl > 50: 200mg c/24hCl 30-49: 200mg c/48hCl 15-29: 200mg c/72hCl < 15: 200mg c/96hEn solución (10 mg/ml):aCl > 50: 240mg (24 ml) c/24hCl 30-49: 120mg (12 ml) c/24hCl 15-29: 80mg (8 ml) c/24hCl <15: 60mg (6 ml) c/24hTruvada©: no administrar a pacientes con Cl > 30 ml/min | HD: en comprimidos 200mg c/96h, en solución (10 mg/ml) 60mg (6ml) c/24hLos días de HD administrar post-HDNo se ha estudiado en diálisis peritoneal.Truvada®: no administrar a pacientes en HD (administrar los componentes por separado, ajustando dosis adecuadamente) | |
| Zidovudina (AZT) | 300 mg/12h | Puede acumularse el metabolito glucurónido (GAZT)Cl 10-50: 250-300mg c/12hCl < 10: 300mg c/24hNo administrar Combivir® y Trizivir® en pacientes con Cl < 50 ml/min (administrar los componentes por separado, ajustando dosis adecuadamente) | 300mg c/24hHD/CAPD: no afecta la eliminación de AZT y aumenta la eliminación de GAZT. Por precaución, se recomienda administrar la dosis diaria post-HD/CAPD | |
| Tenofovir (TDF)Se recomienda valorar riesgo/beneficio del empleo de tenofovir en pacientes con insuficiencia renalLos ajustes han sido obtenidos mediante modelado de datos farmacocinéticos tras la administración de dosis únicas en sujetos con VIH y VHB con diferentes grados de insuficiencia renal, incluyendo HD. Su eficacia y seguridad no han sido evaluadas clínicamente, por lo que se recomienda una estrecha monitorización | 300 mg/24h | Se recomienda emplear la formulación en gránulosCl ≥ 50: no requiere ajuste de dosisCl 30-49: 132mg (4 cacitos)/24hCl 20-29: 65mg (2 cacitos)/24hCl 10-19: 33mg (1 cacito)/24hNo hay recomendaciones disponibles para pacientes con Cl < 10 sin HD | HD: 16,5mg (medio cacito) tras completar cada sesión de HD de 4h | |
| Inhibidores de la transcriptasa inversa no nucleósidos | Efavirenz (EFV) | 600 mg/24h | No requiere ajuste de dosisAtripla®: en pacientes con Cl < 50 ml/min, utilizar los principios activos por separado | HD: no parece necesario ajustar la dosisCAPD: un estudio farmacocinético preliminar indica que no se requiere ajuste de dosis (datos de un solo paciente) |
| Etravirina (ETR) | 200 mg/12h o 400 mg/24h | No requiere ajuste de dosis | HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | |
| Nevirapina (NVP) | 200 mg/12h400 mg/24h (formulación XR) | No requiere ajuste de dosis | HD: los días de HD, se recomienda administrar la dosis después de la HD o un suplemento de 200mg post-HD | |
| Rilpivirina (RPV) | 25 mg/24h | IR leve-moderada: no requiere ajuste de dosisIR grave: no hay datos. Emplear con precaución. La combinación de rilpivirina con un inhibidor potente del CYP3A4 como ritonavir o cobicistat únicamente se debe usar en estos pacientes si el beneficio supera el riesgo | HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | |
| Inhibidores de la proteasa del VIH | Atazanavir (ATV) | 300 mg/24h (con 100 mg/24h de ritonavir)400 mg/24h (sin ritonavir) | No requiere ajuste de dosis | HD/CAPD: por su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPDHD: se recomienda su uso potenciado (ATV/r 300/100) para compensar el descenso de concentración de ATV (reducción del 28% en el AUC de ATV los días sin HD y del 42% los días de HD; la eliminación a través de la HD es de solo el 2%). Monitorizar niveles plasmáticos cuando sea posible |
| Darunavir (DRV) | 800 mg/24h (con 100 mg/24h de ritonavir) | IR leve, moderada o grave: no requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | |
| Fosamprenavir (FPV) | 700 mg/12h (con 100 mg/12h de ritonavir) | No requiere ajuste de dosis | HD/CAPD: debido a su elevada unión a proteínas plasmáticas, no es de esperar que se elimine en las sesiones de HD/CAPD | |
| Indinavir (IDV) | 800 mg/12h (con 100 mg/12h de ritonavir)800 mg/8h (sin ritonavir) | No requiere ajuste de dosis | HD: probablemente no requiera ajuste de dosis si la función hepática está conservada (datos de un solo paciente). Se elimina mínimamente a través de la HD | |
| Lopinavir (LPV/r) | 400/100 mg/12h | No requiere ajuste de dosis | HD: el AUC de LPV/r en 13 pacientes en HD fue equivalente a la de pacientes con función renal normal. No ajuste de dosisCAPD: no hay datos. Debido a la elevada unión a proteínas plasmáticas de lopinavir y ritonavir, no es de esperar que se elimine en las sesiones de CAPD | |
| Ritonavir | Potenciador farmacocinético: dosis variable en función del IP al que acompaña | No requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas de ritonavir, no es de esperar que se elimine en las sesiones de HD/CAPD | |
| Saquinavir (SQV) | 1.000 mg/12h (con 100 mg/12h de ritonavir) | No requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas de ritonavir, no es de esperar que se elimine en las sesiones de HD/CAPD. Datos de un paciente indican escasa eliminación a través de HD | |
| Tipranavir (TPV) | 500 mg/12h (con 200 mg/12h de ritonavir) | No requiere ajuste de dosis | HD/CAPD: debido a la elevada unión a proteínas plasmáticas de TPV/ ritonavir, no es de esperar que se eliminen en las sesiones de HD/CAPD | |
| Inhibidores de la fusión | Enfuvirtida (T-20) | 90 mg/12h (vía s.c.) | No requiere ajuste de dosis | HD: no requiere ajuste de dosis |
| Inhibidores de la integrasa | Raltegravir (RAL) | 400 mg/12h | No requiere ajuste de dosis | HD: no es probable que RAL se elimine significativamente a través de la HD. Datos de 2 pacientes con ERCA mostraron la ausencia de eliminación de RAL durante una sesión de HD de 4h |
| Dolutegravir (DTG) | 50 mg/24h | No se requiere ajuste de dosis. Se realizó un estudio sobre la farmacocinética de dolutegravir en sujetos con insuficiencia renal grave (Cl<30 ml/min) emparejados con controles sanos. La exposición a dolutegravir disminuyó aproximadamente un40% en sujetos con insuficiencia renal grave. Se desconoce el mecanismo. No se considera necesario ningún ajuste de dosis, pero se recomienda precaución en pacientes con IR avanzada y experiencia previa a INI que presenten mutaciones o sospecha de resistencia a estos fármacos. In vitro, dolutegravir inhibe los transportadores renales OCT2 y MATE1. Aunque in vitro se ha observado inhibición de OAT1B1 y OAT1B3, no parece que in vivo tenga repercusión clínica.b Los transportadores OCT2 y MATE1 intervienen en la secreción renal de creatinina, por lo que se ha observado una disminución del aclaramiento de creatinina durante las primeras 4 semanas del 10-14%, sin que ello esté relacionado con un empeoramiento del filtrado glomerular | No se dispone de datos, aunque no se esperan diferencias farmacocinéticas en esta población | |
| Elvitegravir (EVG)/cobicistat/TDF/FTC | 150/150/200/245 mg | Cobicistat reduce levemente el filtrado glomerular renal estimado de creatinina (aunque no altera el filtrado glomerular real) debido a la inhibición de la secreción tubular de creatininaLa asociación EVG/COBI7TDF/FTC no debe iniciarse en pacientes con Cl <70 ml/min. Si el Cl durante el tratamiento se reduce a Cl <70 ml/min, monitorizar estrechamente la función renal (ver ficha técnica de Stribild®); si se reduce a Cl<50 ml/min debe suspenderse, dado que no se puede realizar el ajuste de dosis adecuado para TDF/FTC | La combinación EVG/COBI7TDF/FTC no debe emplearse si Cl <50 ml/min | |
| Antagonistas del correceptor CCR5 | MVR (maraviroc) | Dosis variable en función de los antirretrovirales asociados, consultar ficha técnicaEn general, 150 mg/12h siempre que estén presentes inhibidores potentes del CYP3A4 como los IP/r (excepto TPV/r)300 mg/12h junto con TPV/r, ITIAN, NVP y RAL600 mg/12h junto con inductores importantes del CYP3A4, como EFV | En ausencia de inhibidores potentes del CYP3A4 no requiere ajuste de dosisSolo se recomienda un ajuste de dosis en pacientes con Cl <80 ml/min y que están recibiendo inhibidores potentes del CYP3A4, como los IP/r (excepto TPV/r), cobicistat, ketoconazol, itraconazol, voriconazol, claritromicina, telitromicina, boceprevir o telaprevir: en estos casos administrar 150mg c/24h. Si el Cl es <30 ml/min se recomienda mucha precaución debido al aumento de riesgo de hipotensión posturalCon Cl <80 ml/min y en combinación con FPV/r, administrar 150mg c/12hCon Cl <80 ml/min y en combinación con TPV/r no se requiere ajuste de dosis (300mg c/12h)(Estos ajustes de dosis se recomiendan basándose en los datos de un estudio en insuficiencia renal y simulaciones farmacocinéticas, sin que su seguridad y eficacia hayan sido evaluadas clínicamente, por lo que se recomienda una estrecha monitorización) | HD: en ausencia de inhibidores potentes del CYP3A4 no se requiere ajuste de dosis. En presencia de los mismos, dosificar igual que para Cl <80 ml/min (datos limitados)Escasa eliminación a través de la HD |
CH: cirrosis hepática; Cl: aclaramiento de creatinina en ml/min; ERCA: enfermedad renal crónica avanzada, HD: hemodiálisis; IH: insuficiencia hepática; IP/r: inhibidores de la proteasa potenciados con ritonavir; IR: insuficiencia renal; MATE1: transportador de expulsión de toxinas y multifármacos 1; OAT1 y 3: transportadores de aniones orgánicos 1 y 3; OCT2: transportador de cationes orgánicos 2.
- –
Es necesario ajustar las dosis de los ITIAN, excepto en el caso de ABC (A-II).
- –
No se requiere ajuste de dosis para los ITINN, IP, ENF, ni los INI RAL y DTG (A-II).
- –
MVC requiere ajuste de dosis si se emplea en combinación con inhibidores potentes del CYP3A4, como los IP (excepto TPV/r), ketoconazol, itraconazol, claritromicina y telitromicina (A-II).
- –
Se desaconseja el uso de coformulaciones a dosis fijas de FAR en pacientes con insuficiencia renal significativa. En estos casos deben emplearse los FAR por separado y realizar los ajustes pertinentes (B-III).
- –
En los pacientes con cualquier grado de insuficiencia renal se recomienda vigilar estrechamente la función renal y evitar los fármacos potencialmente nefrotóxicos (A-III).
- –
En los pacientes con enfermedad renal crónica avanzada se debe realizar el ajuste de dosis recomendado por las fichas técnicas de cada medicamento, teniendo en cuenta las posibles interacciones entre los diferentes fármacos, más frecuentes y peligrosas en esta situación (A-II).
• Momento de inicio del TAR. El control de la replicación del VIH-1 y la mejoría inmunológica por efecto del TAR se han asociado a una menor velocidad de progresión de la enfermedad hepática causada por VHC y a una mejor evolución clínica, incluso en pacientes con cirrosis descompensada, en estudios de cohortes227,228. Por ello, el TAR es una prioridad en estos pacientes.
En los coinfectados en los que se plantea el tratamiento del VHC, en general es preferible iniciar el TAR y, una vez controlada la replicación del VIH-1, añadir el tratamiento de la hepatitis. Opcionalmente, sobre todo en pacientes controladores de elite, se podría plantear tratar inicialmente el VHC.
TDF suprime el VHB en la inmensa mayoría de los pacientes coinfectados por VIH-1 y VHB229. Por tanto, si existe indicación de tratar la infección por VHB, se debe iniciar el TAR con un régimen que incluya TDF.
GeSIDA ha elaborado unas guías de manejo de las hepatitis virales en pacientes con infección por VIH-1, que prontamente estarán disponibles.
Recomendaciones- –
En pacientes coinfectados por el VHC se recomienda iniciar el TAR, independientemente de la cifra de linfocitos CD4+ (A-II).
- –
En pacientes que precisan tratamiento de la hepatitisC, en general es preferible iniciar el TAR (A-III).
- –
En pacientes coinfectados por VIH-1 y VHB con indicación de tratamiento frente al VHB, se debe iniciar el TAR con un régimen que incluya TDF (A-I).
• Elección de los FAR. La elección de los FAR en un paciente coinfectado por virus de la hepatitis ha de tener en cuenta su potencial hepatotoxicidad, la existencia de cirrosis hepática, la coinfección por VHB y la indicación de tratamiento anti-VHC.
La hepatotoxicidad asociada al TAR es más frecuente en pacientes coinfectados. Sin embargo, si se exceptúan los casos asociados a dideoxinucleósidos (d-AN) y NVP, que pueden causar fallo hepático agudo y toxicidad crónica, los FAR utilizados actualmente se asocian con un bajo riesgo de hepatotoxicidad y la mayoría de los episodios de hepatitis tóxica por TAR son leves230.
Por otra parte, no se ha demostrado de modo consistente que ningún FAR ejerza un efecto protector sobre la esteatogénesis ni la fibrogénesis hepática.
En pacientes con cirrosis claseA de Child-Pugh se puede usar cualquier FAR, con las consideraciones antes expuestas. En pacientes con insuficiencia hepatocelular (clasesB y C de Child-Pugh) vale la pena tener en cuenta que, con respecto a los ITINN, los IP/r presentan un mayor margen terapéutico. Los datos farmacocinéticos obtenidos con FPV permiten la dosificación individualizada en pacientes con cirrosis Child-PughB y C231.
La monitorización de los niveles plasmáticos, teóricamente útil en pacientes con insuficiencia hepatocelular, no está disponible en la mayoría de los centros y su utilidad en el tratamiento de los pacientes con insuficiencia hepática no se ha demostrado.
Por sus características farmacocinéticas, los INI ofrecen ventajas en estos pacientes. RAL ha demostrado niveles séricos adecuados, sin necesidad de ajuste de dosis y buena tolerabilidad en pacientes en estadioC de Child232. En base a los datos obtenidos en voluntarios sanos, DTG tampoco precisa ajuste de dosis en caso de insuficiencia hepática moderada233. En los pacientes que reciben tratamiento anti-VHC con RBV están contraindicados d4T y ZDV porque aumentan el riesgo de toxicidad. Además, existen múltiples interacciones entre los FAR y los antivirales de acción directa (AAD) frente al VHC, excepto sofosbuvir, que condicionan la modificación de la dosis o desaconsejan su coadministración234-239 (tabla 14).
Interacciones más relevantes desde el punto de vista clínico entre fármacos antirretrovirales y antivirales directos frente al VHC
| Boceprevir (BOC) 800 mg/QD | Telaprevir (TLV) 750 mg/TID | Simeprevir (SMV) 150 mg/QD | Daclatasvir (DCV) 60 mg/QD | Sofosbuvir (SOFOS) 400 mg/QD | |
|---|---|---|---|---|---|
| TDF300 mg/QD | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-I)SMV aumenta los niveles de TDF. Vigilar nefrotoxicidad (A-I) | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-I)TDF: aumenta la Cmáx 25% |
| ATV/r300/100 mg/QD | Considerar coadministración, controlado la eficacia del TAR (B-I)ATV: Cmín disminuye: 49% | No precisa ajuste de dosis. Vigilar niveles de bilirrubina (A-I)ATV: Cmín aumenta 85% | No recomendado (A-III) | Daclatasvir (DCV), 30 mg/QD (A-I)DCV: Cmín aumenta > 2 vecesAUC GMR (IC 90%: 2,10; 1,95-2,26) | No precisa ajuste de dosis (A-III) |
| DRV/r800/100 mg/QD | No recomendado (A-I)DRV: Cmín disminuye 59%BOC: Cmín disminuye 35% | No recomendado (A-I)DRV: Cmín disminuye 58%TLV: Cmín disminuye 32% | No recomendado (A-I)DRV: Cmín aumenta 31%SMV: Cmín aumenta 5 veces | No precisa ajuste de dosis (A-I)AUC GMR (IC 90%: 1,41; 1,32-1,50 | No precisa ajuste de dosis (A-I)SOFOS: AUC aumenta un 45% |
| FPV/r700/100/BID | No recomendado (A-III) | No recomendado (A-I)FPV: Cmín disminuye 56%TLV: Cmín disminuye 30% | No recomendado (A-III) | Interacción no estudiada | No precisa ajuste de dosis (A-III) |
| LPV/r400/100 mg/QD | No recomendado (A-I)LPV: Cmín disminuye 43%BOC: Cmín disminuye 57% | No recomendado (A-I)TLV: Cmín disminuye 52% | No recomendado (A-III) | Interacción no estudiada | No precisa ajuste de dosis (A-III) |
| EFV600 mg/QD | No recomendado (A-I)BOC: Cmín disminuye 44% | TLV 1125 mg/TID (A-I)TLV: Cmín disminuye 47% | No recomendado (A-I)SMV: Cmín disminuye 91% | DCV 90 mg/QD (A-I)DCV: Cmín disminuye 59%AUC GMR (IC 90%:0,68; 0,68-0,78) | No precisa ajuste de dosis (A-I) |
| ETV200mg/BID | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-I) | No recomendado (A-III) | Interacción no estudiada, no se recomienda su uso | No precisa ajuste de dosis (A-III) |
| RPV25 mg/QD | No precisa ajuste de dosis (A-I)RPV: Cmín aumenta 51% | No precisa ajuste de dosis (A-I)RPV: Cmín aumenta 1,9 veces | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-I) |
| DTV50 mg/QD | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-III) |
| EVG/COBI/FTC/TDF150 mg/150 mg/200 mg/245 mg/QD | No recomendado (A-III) | No precisa ajuste de dosis (A-I)EVG: Cmín aumenta 32% | No recomendado (A-III) | DCV 30mg QD (A-III) | No precisa ajuste de dosis (A-III) |
| RAL400 mg/BID | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-I) | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-I) |
| MVC300 mg/BID | MVC 150mg BID (A-I)MVC: Cmín aumenta 2,8 veces | MVC 150mg BID (A-I)MVC: Cmín aumenta 10 veces | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-III) | No precisa ajuste de dosis (A-III) |
- –
En pacientes con hepatopatía crónica y función hepática conservada, incluida la cirrosis claseA de Child, se puede utilizar cualquier FAR (A-I), aunque es razonable evitar los dideoxinucleótidos (A-III).
- –
En pacientes con insuficiencia hepatocelular, los INI no precisan ajuste de dosis (A-I). Los IP/r presentan mayor margen terapéutico que los ITINN (A-II).
- –
Debe evitarse la asociación de RBV con ddI, d4T y ZDV (A-I).
- –
Excepto sofosbuvir, los AAD disponibles (telaprevir, boceprevir, simeprevir y daclatasvir) presentan interacciones farmacocinéticas significativas con los ITINN y los IP/r, que requieren ajuste de dosis o contraindican la coadministración (A-I).
Se recomienda la lectura de los 2 documentos realizados por GeSIDA en los que se tratan de forma amplia las neoplasias240,241.
Los pacientes con cáncer que son sometidos a quimioterapia/radioterapia tienen riesgo de inmunodepresión y de disminución del recuento de los linfocitos CD4+, lo que ya de por sí justificaría el inicio de TAR242. En este escenario es importante considerar el perfil de toxicidad y las posibles interacciones farmacocinéticas, principalmente de aquellos FAR que se metabolizan por la vía del CYP450, con los fármacos antineoplásicos243. Los IP/r se asocian a neutropenia más profunda y prolongada cuando se coadministran con la quimioterapia de los linfomas240; algunos de ellos (ATV, LPV y SQV) alargan el intervalo QT, lo que podría potenciar este efecto cuando se usan junto a muchos fármacos anticancerosos244. Además, en un estudio244 presentaron mayor toxicidad, más interacciones y menor eficacia virológica que los ITINAN y RAL.
Recomendaciones- –
El TAR es un pilar fundamental en el tratamiento de los pacientes con infección por el VIH con sarcoma de Kaposi o linfomas no Hodgkin (A-II).
- –
En pacientes con otro tipo de neoplasias, como consideración general, si no realizan TAR, este ha de empezarse tan pronto como sea posible (A-II).
- –
RAL por sus características farmacológicas, su excelente tolerabilidad y las mínimas interacciones, debe ser el FAR de elección en pacientes que reciban quimioterapia, siempre que sea posible (A-III).
El TAR ha reducido la mortalidad relacionada con el sida y ha mejorado la calidad de vida de los pacientes. Sin embargo, su coste es elevado y, en un entorno donde los recursos son limitados, es necesario gestionar adecuadamente el presupuesto. En las diferentes combinaciones de TAR utilizadas como terapia de inicio existen diferencias sustanciales entre ellas: el gasto mensual con pautas de eficacia similar puede diferir en cantidades de más de 500euros.
Una evaluación farmacoeconómica con objeto de determinar el posicionamiento de nuevas estrategias o de nuevos medicamentos debe contemplar no solamente el coste, sino también la eficacia (ensayos clínicos) o la efectividad (práctica clínica habitual) de forma conjunta. Por este motivo, en los últimos años se publica conjuntamente con estas guías un estudio farmacoeconómico245 en el que se realiza una evaluación de costes y eficiencia (coste/eficacia) mediante construcción de árboles de decisión a partir de las pautas preferentes recomendadas.
El precio que el sistema nacional de salud paga por un fármaco se obtiene a partir del precio de venta laboratorio (PVL) con la deducción obligatoria del 7,5% sobre el precio de compra de medicamentos no genéricos y no afectados por el sistema de precios de referencia (Real Decreto-ley 8/2010, de 20 de mayo: Medidas extraordinarias para la reducción del déficit público). En el caso de que hubieran transcurrido 10años desde la fecha de inicio de financiación con fondos públicos (11años en el caso de haber sido autorizada una nueva indicación), la deducción sería del 15%, salvo en los medicamentos que cuenten con protección de patente de producto en todos los estados miembros de la Unión Europea (Real Decreto-ley 9/2011, de 19 de agosto). Sin embargo, ninguno de los FAR incluidos en los regímenes recomendados como terapia de inicio tiene deducción del 15% en noviembre de 2014. A esta cifra habría que sumar el 4% de IVA. Además cabe considerar que pueden existir variaciones entre los precios finales de adquisición entre distintas comunidades autónomas e, incluso, entre distintos hospitales en una misma comunidad.
Por ello, es importante que cada centro utilice sus propios precios para obtener datos de eficiencia. A tal efecto, el estudio mencionado245 ofrece una aplicación informática en la que, introduciendo los precios que cada hospital paga, se obtiene el posicionamiento de las distintas pautas de inicio. La aplicación puede descargarse de forma gratuita en la web de GeSIDA, en Guías clínicas (http://www.gesida-seimc.org/guias_clinicas.php?mn_MP=406&mn_MS=407), en el apartado Costs and cost-efficacy analysis of the GESIDA/Spanish National AIDS Plan recommended guidelines for initial antiretroviral therapy in HIV-infected adults (aplicación cálculo coste y eficacia).
Otra consideración a tener en cuenta es que los estudios farmacoeconómicos probablemente subestimen la efectividad del TAR, dado que habitualmente no incluyen la reducción en el riesgo de transmisión de la enfermedad de los pacientes tratados. Este hecho puede tener un impacto económico importante.
Recomendación- –
Se recomienda incluir criterios de coste-efectividad en la toma de decisiones sobre el TAR de inicio (A-III).
Koldo Aguirrebengoa ha efectuado labores de consultoría en los laboratorios AbbVie, Gilead Sciences y Janssen, ha participado en ensayos y estudios clínicos de AbbVie, MSD, Gilead, Janssen y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
José R. Arribas ha efectuado labores de consultoría en los laboratorios AbbVie Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, Tobira y ViiV Healthcare, ha disfrutado de becas para investigación clínica de Janssen, MSD y Gilead, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Juan Berenguer ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead, Janssen-Cilag, Janssen Therapeutics, Merck y ViiV Healthcare, ha disfrutado de becas para investigación clínica de Bristol-Myers Squibb, Merck Sharp & Dohme y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare.
José R. Blanco ha efectuado labores de consultoría en los laboratorios AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck y ViiV Healthcare, ha recibido compensación económica por charlas de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Janssen, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Gilead Sciences y Bristol-Myers Squibb.
Vicente Boix ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Boehringer-Ingelheim, Gilead Sciences, Janssen, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare
José Luis Casado ha efectuado labores de consultoría para AbbVie y ViiV Healthcare; ha recibido compensaciones económicas por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare, así como ha disfrutado de becas para investigación clínica de Janssen, Gilead Sciences y ViiV Healthcare.
Bonaventura Clotet ha efectuado labores de consultoría o participado en ensayos clínicos o en charlas retribuidas con los siguientes laboratorios farmacéuticos: BMS, AbbVie, Gilead, Janssen, Merck (MSD) y ViiV Healthcare
Manuel Crespo ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, ha disfrutado de becas para investigación clínica de laboratorios Gilead Sciences y ViiV Healthcare, ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, así como pagos por desarrollo de presentaciones educacionales para Bristol-Myers Squibb, Gilead Sciences y ViiV Healthcare.
Pere Domingo ha efectuado labores de consultoría en los laboratorios AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare, ha disfrutado de becas para investigación clínica de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare.
Vicente Estrada ha efectuado labores de consultoría en los laboratorios AbbVie, Gilead Sciences y Janssen; ha disfrutado de becas para investigación clínica de AbbVie, Boehringer-Ingelheim, Gilead Sciences y Janssen, y ha recibido compensación económica por charlas de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Federico García ha efectuado labores de consultoría y ha recibido compensación económica por charlas en los laboratorios Gilead Sciences, ViiV, AbbVie y Roche Diagnostica.
José M. Gatell ha efectuado labores de consultoría en los laboratorios AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare, ha disfrutado de becas para investigación clínica de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare.
Juan González García ha efectuado labores de consultoría en los laboratorios Abbott Laboratories, AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Félix Gutiérrez ha efectuado labores de consultoría en los laboratorios Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas y/o preparación de material docente de Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare.
José Antonio Iribarren ha efectuado labores de consultoría en los laboratorios AbbVie y Janssen-Cilag, ha recibido becas de investigación clínica de laboratorios AbbVie, Bristol-Myers Squibb, Gobierno Vasco, FIPSE y FISS, ayudas para asistencia a Congresos de AbbVie, Gilead, Janssen-Cilag y ViiV, y ha participado en actividades educativas, charlas o simposios patrocinados por AbbVie, Bristol-Myers Squibb, Gilead, Merck, Novartis Janssen, Pfizer y ViiV.
Hernando Knobel ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, y ha recibido compensaciones económicas por el desarrollo de presentaciones educacionales para AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Josep M. Llibre ha efectuado labores de consultoría para Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare, y ha recibido compensación económica por actividades de formación de Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare.
Jaime Locutura ha efectuado labores de consultoría para AbbVie, Bristol-Myers Squibb, Gilead Sciences y ViiV Healthcare, ha recibido compensaciones económicas por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen-Cilag, Merck Sharp & Dohme y ViiV Healthcare, así como pagos por desarrollo de material educacional para AbbVie, Boehringer-Ingelheim y ViiV Healthcare.
José López Aldeguer ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen y ViiV Healthcare, ha disfrutado de becas para investigación clínica de Bristol-Myers Squibb y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Juan Carlos López Bernaldo de Quirós ha efectuado labores de consultoría para AbbVie, Bristol-Myers Squibb, Boehringer-Ingelheim, Gilead Sciences, Janssen, Merck-Sharp & Dome, Roche Pharmaceuticals y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Boehringer-Ingelheim, Gilead Sciences, Janssen, Merck-Sharp & Dome, Roche Pharmaceuticals y ViiV Healthcare. También ha recibido pagos por desarrollo de material educacional para Boehringer-Ingelheim, Gilead Sciences y ViiV Healthcare.
Fernando Lozano ha efectuado labores de consultoría para AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck-Sharp & Dome y ViiV Healthcare, y ha recibido compensación económica por charlas con fines educacionales de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck-Sharp & Dome y ViiV Healthcare.
José M. Miró ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck, Novartis y Sanofi, ha disfrutado de becas para investigación clínica de Cubist, Gilead, ViiV, Novartis, Merck, Fondo de Investigaciones Sanitarias (FIS) del Instituto de Salud Carlos III (Madrid), Fundación para la Investigación y Prevención del Sida en España (FIPSE, Madrid), Ministerio de Sanidad, Servicios Sociales e Igualdad (MSSSI, Madrid), National Institutes of Health (NIH, Bethesda, MA, EE.UU.) y NEAT, y ha recibido compensación económica por charlas de los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck, Novartis y ViiV Healthcare.
Santiago Moreno ha efectuado labores de consultoría en los laboratorios AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y Roche; ha disfrutado de becas para investigación clínica de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y Roche, y ha recibido compensación económica por charlas de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y Roche.
Daniel Podzamczer ha efectuado labores de consultoría en los laboratorios AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare; ha disfrutado de becas para investigación clínica de Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck y ViiV Healthcare.
Rosa Polo declara no tener conflicto de intereses.
Joaquín Portilla ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, ha disfrutado de becas para investigación clínica de AbbVie, Janssen, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Federico Pulido ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare, y ha recibido compensación económica por charlas de Abbott Laboratories, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck y ViiV Healthcare.
Esteban Ribera ha realizado asesorías o ha recibido ayudas para investigación o docencia en relación con la infección por el VIH de los siguientes laboratorios farmacéuticos: AbbVie, BMS, Ferrer International, Gilead, GSK, Janssen-Cilag, MSD, Pfizer, Roche Farma, Schering Plough y ViiV.
Melchor Riera ha recibido ayudas para viajes a reuniones y congresos de Janssen, Merck y Gilead Sciences, ha recibido compensación económica por charla de AbbVie, y aportaciones económicas al servicio para labores docentes o de investigación de laboratorios AbbVie y Bristol-Myers Squibb.
Antonio Rivero ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck (MSD) y ViiV Healthcare; ha disfrutado de becas para investigación clínica de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck (MSD) y ViiV Healthcare, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Merck (MSD) y ViiV Healthcare.
Rafael Rubio ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences y Janssen, y ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, Roche y ViiV Healthcare.
Jesús Santos ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences y Janssen, ha recibido compensación económica por charlas de Bristol-Myers Squibb, MSD, Janssen y Gilead Sciences, así como pagos por desarrollos de presentaciones educacionales para Bristol-Myers Squibb, Gilead Sciences, Janssen y MSD.
José Sanz Moreno ha participado en ensayos clínicos promovidos por Bristol-Myers Squibb y ViiV Healthcare, y ha desarrollado labores de consultoría e impartido presentaciones con fines docentes, organizadas por AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck Sharp & Dohme y ViiV Healthcare, por las que ha percibido compensación económica.
Jesús Sanz ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, ViiV Healthcare y Boehringer-Ingelheim, ha recibido compensación económica por charlas de AbbVie, Bristol-Myers Squibb, Gilead Sciences, Janssen, Merck, ViiV Healthcare y Boehringer-Ingelheim, y ha recibido pagos por desarrollo de presentaciones educacionales para ViiV Healthcare.
María Jesús Téllez ha efectuado labores de consultoría en los laboratorios AbbVie, Bristol-Myers Squibb, Gilead Sciences y Janssen, y ha recibido compensación económica por charlas de Gilead Sciences y Janssen.
Montserrat Tuset ha disfrutado de becas para investigación clínica de laboratorios Bristol-Myers Squibb, Gilead Sciences, Merck y Janssen, y ha recibido compensación económica por charlas de Janssen, Merck, Gilead y ViiV Healthcare.
La Junta Directiva de GeSIDA y el Plan Nacional sobre el Sida agradecen las aportaciones y opiniones de: Esther Cabrero, Manuel Cotarelo, Jorge del Romero, Carlos Dueñas, Pedro Ferrer, Beatriz Hernández Novoa, Henar Hevia, Luis Fernando López Cortés, Cristina Loriente, Juan Emilio Losa García, José Emilio Martín Herrero, José Antonio Pérez Molina, Felipe Rodríguez Alcántara, Nuria Sánchez, Óscar Serrano y María Eugenia Vispo.
Juan Berenguer, Hospital General Universitario Gregorio Marañón, Madrid.
Rosa Polo, Secretaría del Plan Nacional sobre el Sida, Ministerio de Sanidad, Servicios Sociales e Igualdad.
Antonio Rivero, Hospital Universitario Reina Sofía/IMIBIC, Córdoba.
José López Aldeguer, Hospital Universitario La Fe, IIS La Fe, Valencia.
Fernando Lozano, Hospital Universitario Virgen de Valme, Sevilla.
Koldo Aguirrebengoa, Hospital Universitario de Cruces, Bilbao.
José Ramón Arribas, Hospital Universitario La Paz/IdiPAZ, Madrid.
José Ramón Blanco, Hospital San Pedro, Logroño.
Vicente Boix, Hospital General Universitario de Alicante.
José Luis Casado, Hospital Ramón y Cajal/IRYCIS, Madrid.
Bonaventura Clotet, Hospital Germans Trias i Pujol, Badalona, Barcelona.
Manuel Crespo, Hospital Vall d’Hebron, Barcelona.
Pere Domingo, Hospital de la Santa Creu i Sant Pau, Barcelona.
Vicente Estrada, Hospital Clínico San Carlos, Madrid.
Federico García, Complejo Hospitalario Universitario de Granada, Granada.
José María Gatell, Hospital Clínic/IDIBAPS, Universidad de Barcelona, Barcelona.
Juan González García, Hospital Universitario La Paz/IdiPAZ, Madrid.
Félix Gutiérrez, Hospital General Universitario de Elche, Universidad Miguel Hernández, Alicante.
José Antonio Iribarren, Hospital Universitario Donostia, San Sebastián.
Hernando Knobel, Hospital del Mar, Barcelona.
Josep Maria Llibre, Hospital Germans Trias i Pujol, Badalona, Barcelona.
Jaime Locutura, Hospital Universitario, Burgos.
Juan Carlos López, Hospital General Universitario Gregorio Marañón, Madrid.
José M. Miró, Hospital Clínic/IDIBAPS, Universidad de Barcelona, Barcelona.
Santiago Moreno, Hospital Ramón y Cajal/IRYCIS, Madrid.
Daniel Podzamczer, Hospital Universitari de Bellvitge, L’Hospitalet, Barcelona.
Joaquín Portilla, Hospital General Universitario, Alicante.
Federico Pulido, Hospital Universitario 12 de Octubre/i+12, Madrid.
Esteban Ribera, Hospital Vall d’Hebron, Barcelona.
Melchor Riera, Hospital Son Espases, Palma de Mallorca.
Rafael Rubio, Hospital Universitario 12 de Octubre/i+12, Madrid.
Jesús Santos, Hospital Universitario Virgen de la Victoria, Málaga.
José Sanz Moreno, Hospital Universitario Príncipe de Asturias, Alcalá de Henares, Madrid.
Jesús Sanz Sanz, Hospital Universitario de la Princesa, Madrid.
María Jesús Téllez, Hospital Clínico San Carlos, Madrid.
Montserrat Tuset, Hospital Clínic/IDIBAPS, Universidad de Barcelona, Barcelona.

