









Introducción
Las hepatitis víricas forman un conjunto de patologías de origen infeccioso de enorme repercusión en la práctica clínica y en la salud pública. Las formas agudas suelen cursar en forma autolimitada y curar sin secuelas, aun cuando pueden presentarse como cuadros de hepatitis fulminante y ser causa de muerte por insuficiencia hepática aguda. Las formas crónicas son, si embargo, las más importantes desde cualquier punto de vista, ya que se prolongan de por vida y evolucionan con moderada frecuencia hacia la cirrosis hepática y el carcinoma primario de hígado, y constituyen la causa más frecuente para el trasplante hepático. El hecho de que la sangre de los pacientes que albergan estas infecciones crónicas sea un vehículo muy eficaz para la transmisión de los agentes involucrados añade, además, otros elementos que incrementan su importancia. Así, las hepatitis víricas en receptores de sangre y hemoderivados han condicionado el mundo de la transfusión sanguínea y de la hemoterapia desde hace décadas, obligando a la adopción de estrictas medidas para la selección de los donantes y para el control de las hemodonaciones en los bancos de sangre y en la industria dedicada al fraccionamiento del plasma. La transmisión nosocomial de esos agentes constituye, hoy día, una preocupación seria en el medio asistencial y un problema aún no resuelto en su totalidad. La diseminación de esos virus entre los usuarios de fármacos de administración parenteral ha aumentado, además, su prevalencia global en las últimas décadas y constituye aún una amenaza para una parte de la población más joven del mundo desarrollado.
La trascendencia del problema que plantean las hepatitis víricas ha sido, así mismo, motor de importantes desarrollos recientes en el terreno de las enfermedades infecciosas. La vinculación directa del virus de la hepatitis B con el cáncer de hígado demostró, por primera vez, la existencia de tumores de origen infeccioso. El uso de los interferones para el tratamiento de las formas crónicas fue pionero en el terreno de la terapia antiviral y la aprobación de la vacuna recombinante frente a la hepatitis B marcó un hito en las aplicaciones médicas de la biotecnología y permitió, por primera vez, disponer de una vacuna capaz de prevenir un cáncer. Pero, además, el estudio de los mecanismos que permiten a algunos de estos virus persistir durante años en una parte de los pacientes a los que infectan está mejorando día a día nuestra comprensión de las complejas interacciones que gobiernan la relación entre el sistema inmunitario y los agentes infecciosos, lo que redundará, a buen seguro, en un conocimiento más profundo del fenómeno global de la infección y la enfermedad infecciosa.
Etiología de las hepatitis víricas
En sentido estricto, las hepatitis víricas responden a infecciones producidas por cinco virus humanos diferentes y filogenéticamente alejados entre sí, que se conocen como virus de la hepatitis A, B, C, D y E (VHA, VHB, VHC, VHD y VHE). En todos los casos, los hepatocitos constituyen sus células hospedadoras principales y las dianas últimas de la infección, si bien son capaces de infectar otras células. En un sentido más amplio, incluyen también cuadros agudos de enfermedad hepática debidos a otros virus humanos no específicamente hepatotropos, pero que pueden producir dichos cuadros como una complicación de la infección 1 (tabla 1). Los virus herpes, en especial el citomegalovirus humano y el virus de Epstein-Barr, son los más frecuentemente involucrados, pero se han descrito hepatitis agudas asociadas a infecciones por virus herpes simple 2, virus de la varicela-zóster 3, virus de la rubéola 4, parvovirus humano B19 5 y adenovirus 6, en ocasiones con presentación fulminante. Además, la afectación hepática es característica de dos infecciones de enorme trascendencia en las regiones tropicales, que pueden presentarse fuera de ellas como casos importados: la fiebre amarilla y la fiebre de dengue. Los agentes conocidos como GBV-C (virus de la hepatitis G, VHG) y virus TT (TTV), inicialmente relacionados con la producción de hepatitis, no han podido confirmarse aún como agentes etiológicos de hepatitis aguda o crónica en el hombre 7,8, aunque el primero se encuentra en asociación con casos esporádicos de hepatitis fulminante 9. Actualmente, se investiga una posible relación entre el circovirus humano conocido como virus SEN y las hepatitis agudas y crónicas 10.

Virus de la hepatitis de transmisión fecal-oral. Virus de la hepatitis A y E
Los VHA y VHE son dos agentes no emparentados filogenéticamente que se transmiten por la vía fecal-oral y causan cuadros autolimitados de hepatitis aguda. Ambos son virus con genoma de ARN monocatenario lineal, no fragmentado y de sentido positivo.
La infección por estos agentes se adquiere por la ingestión de agua o alimentos frescos contaminados o por contacto con las heces de los pacientes que sufren la infección aguda. La puerta de entrada es, esencialmente, la mucosa intestinal y los hepatocitos resultan infectados por la llegada de virus procedente de la viremia primaria. Los síntomas de hepatitis aguda aparecen, cuando lo hacen, tras una media de 3-4 semanas después de adquirida la infección. Las formas fulminantes de la enfermedad aguda son infrecuentes. No obstante, la hepatitis E aguda en mujeres embarazadas presenta una elevada incidencia de casos de presentación fulminante, con tasa de mortalidad elevada. Las infecciones por VHA y VHE no cursan nunca hacia la persistencia, por lo que estos agentes no pueden causar enfermedad hepática crónica.
El VHA es el único miembro conocido del género Hepatovirus dentro de los Picornaviridae, una familia a la que pertenecen los virus de la poliomielitis y otros enterovirus de relevancia en salud. La partícula es desnuda y de simetría icosaédrica, con una cápside formada por cuatro proteínas estructurales. Dos de ellas (VP1 y VP3) poseen los epítopos involucrados en la inducción de la respuesta inmunitaria neutralizante. Las cepas de VHA no muestran diversidad antigénica en dichos epítopos, por lo que los anticuerpos inducidos por cualquier cepa confieren una protección universal frente a la infección. Este hecho ha permitido desarrollar una vacuna inactivada que la previene eficazmente, aunque su uso generalizado en el contexto de una política de eliminación del virus es objeto de controversia.
El genoma del VHA es un ARN monocatenario de 7,5 kb que se organiza en cinco regiones, dos regiones reguladoras en los extremos y tres codificantes en el interior. Tras su desencapsidación, el genoma actúa como mensajero y se traduce en una poliproteína que genera 11 proteínas tras ser procesada por proteasas. Cuatro de ellas (VP1-4) se ensamblan en la cápside de la partícula y las siete restantes son componentes funcionales que participan en la replicación del virus, incluyendo una ARN polimerasa dependiente de ARN. La variabilidad genética de este genoma ya se ha estudiado en profundidad y revela la existencia de no menos de cinco genotipos diferentes 11, aunque su definición se ve dificultada por la frecuente detección de cepas recombinantes. La propuesta de máxima diversidad clasifica las cepas del VHA en siete genotipos (I-VII), y diferencia genosubtipos dentro de los genotipos I (IA, IB) y III (IIIA, IIIB). Sólo los genotipos I, II, III y VII se han encontrado en seres humanos. De ellos, el genotipo I es el más frecuente a escala mundial, aunque el genotipo III no es raro en Europa, incluyendo España 12.
El VHE es un virus humano aún sin clasificar que presenta semejanzas con los miembros de las familias Caliciviridae y Togaviridae13. En los árboles filogenéticos, las secuencias de los genomas del VHE obtenidos de diferentes partes del mundo se agrupan cercanas a las de los Togaviridae, más en concreto a las del virus de la rubéola, y se alejan claramente de la de los Caliciviridae, lo que excluye su pertenencia a esta última familia. La partícula carece, sin embargo, de envoltura, y su cápside parece estar formada por una única proteína. No se conocen variantes antigénicas en el VHE, por lo que el espectro de la inmunidad adquirida tras la infección aguda debe de ser amplio, aunque se sospecha que la protección pueda ser menos duradera de lo que es usual en los virus antigénicamente monotípicos.
El genoma del VHE es, también, un ARN monocatenario de unas 7,2 kb de longitud con capacidad para actuar como mensajero tras su liberación en la célula infectada. Se organiza en dos regiones reguladoras situadas en los extremos y tres marcos abiertos de lectura en fase (open reading frame, ORF), uno largo situado en 59 (ORF1) y dos cortos situados en 39 (ORF2, ORF3). El primero codifica cuatro actividades enzimáticas relacionadas con la replicación del virus y una quinta proteína de función desconocida. El segundo codifica la proteína estructural de la cápside. El tercero, muy corto, codifica un polipéptido de sólo 123 aminoácidos de longitud cuya función es desconocida. El producto del ORF2 contiene los epítopos inmunogénicos principales y podría ser la base de una futura vacuna recombinante frente al VHE.
Se han descrito hasta el momento cuatro genotipos para el VHE. En los análisis filogenéticos, estos cuatro genotipos se agrupan entre sí y se separan de las secuencias del VHE aviar, un virus de reciente descripción 14. Los genotipos I y IV son prevalentes en Asia y en el norte de África, y el genotipo II lo es en América Central. Las cepas aisladas de pacientes del Mediterráneo europeo, incluidos algunos pacientes españoles 15,16, y de Estados Unidos pertenecen al genotipo III, el único encontrado hasta ahora en animales de granja, especialmente en cerdos. Se piensa, por tanto, que en los países desarrollados la hepatitis E podría ser una zoonosis de muy baja incidencia, vinculada quizás al contacto con el ganado porcino.
Virus de la hepatitis de transmisión parenteral
Virus de la hepatitis B
El VHB es un miembro de la familia Hepadnaviridae encuadrado en el género Orthohepadnavirus. Los virus de este género presentan un rango de huésped restringido a mamíferos, mientras que los del género Avihepadnavirus se restringen a aves. El único huésped natural del VHB es el hombre, aunque los chimpancés pueden infectarse de forma natural a partir de portadores humanos.
Los viriones del VHB son esféricos y envueltos, de un diámetro de 40-45 nm. La cápside es icosaédrica y está constituida por una única proteína, que se conoce como antígeno core y que se abrevia como HBcAg. Dentro de ella se encierra el genoma viral y una ADN polimerasa codificada por el propio virus que presenta actividades de transcriptasa inversa, ribonucleasa H y ADN polimerasa dependiente de ADN. La envuelta, de naturaleza lipoproteica, proviene de las membranas del retículo endoplásmico de la célula infectada y presenta tres glucoproteínas de superficie que se anclan en ella y se proyectan hacia el exterior de la partícula. La proteína mayoritaria o antígeno de superficie (proteína S, gp27 o HBsAg) es la más pequeña. En menor proporción, existen otras dos proteínas, más grandes que la anterior: la proteína mediana (proteína M, gp36 o antígeno pre-S2) y la proteína grande (proteína L, gp42 o antígeno pre-S1). Tradicionalmente, el virión del VHB se ha conocido como partícula de Dane, en honor a su descubridor.
Además de los viriones completos, los hepatocitos infectados por el VHB producen en gran cantidad otras estructuras que carecen de ADN y que están formadas exclusivamente por las glucoproteínas de superficie. Las más abundantes consisten en agregados esféricos de 22 nm de diámetro que contienen exclusivamente el HBsAg. En mucha menor proporción, se excretan también agregados filamentosos, del mismo diámetro y de longitud variable, que contienen, también, los antígenos pre-S1 y pre-S2. Esos agregados están presentes en concentraciones muy elevadas en la sangre de las personas infectadas y tienen un papel crucial en la relación con el sistema inmunitario y en el establecimiento de la infección persistente.
Por último, la célula infectada produce también otra proteína vírica que se excreta al torrente circulatorio en forma no agregada. Se trata del antígeno e del VHB (HBeAg) y contiene toda la secuencia del HBcAg más una secuencia agregada al extremo aminoterminal. Esta proteína también tiene un papel muy importante en la patogenia de la infección.
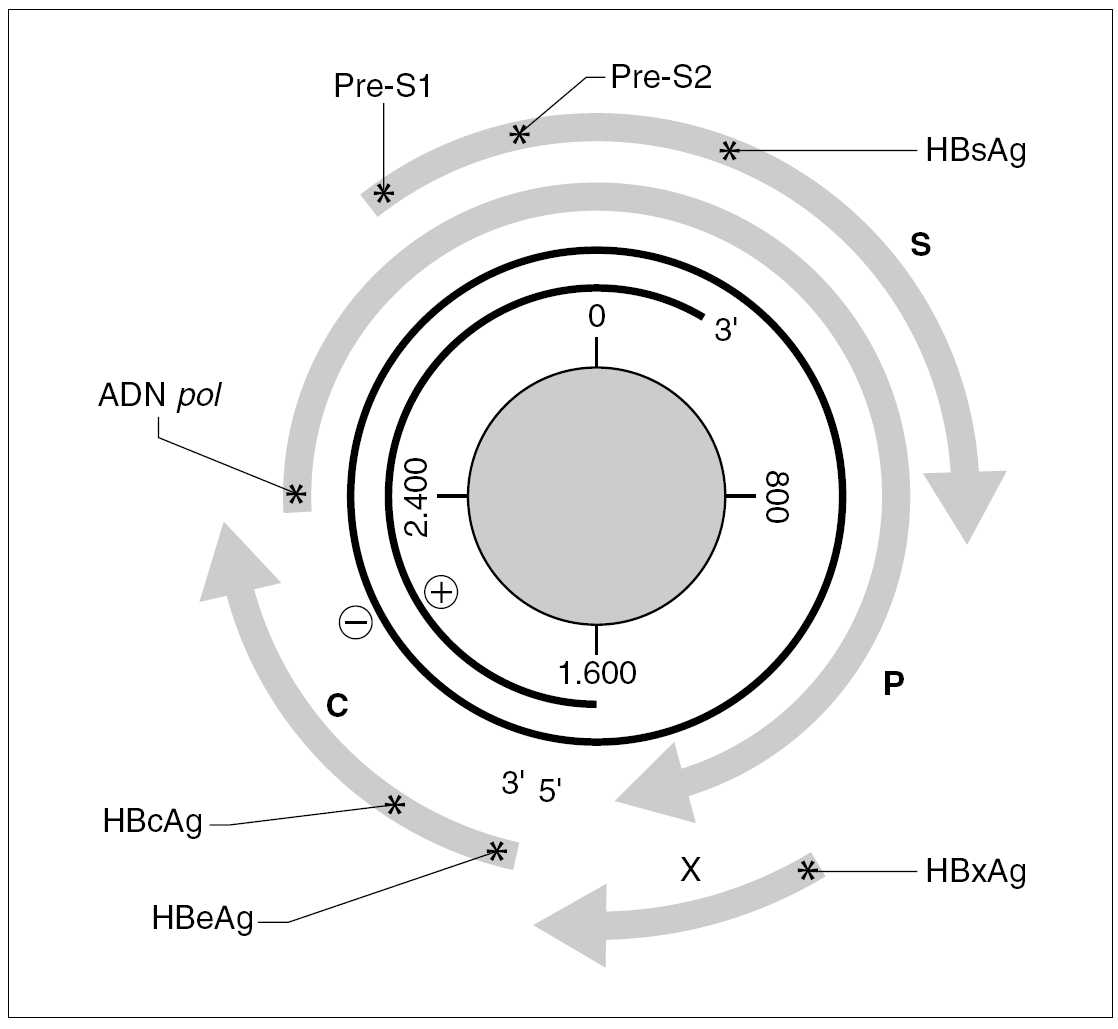
El genoma del VHB consiste en una molécula circular de ADN bicatenario de 3,2 kb, cuya cadena positiva se halla más o menos incompleta en su extremo 39, por lo que el resultado es un ADN no covalentemente cerrado. Este pequeño genoma contiene siete señales de iniciación de la trascripción que definen genes parcialmente solapantes, lo que le confiere una capacidad de codificar proteínas muy superior a la que cabría esperar para su tamaño. Atendiendo a su organización, se distinguen cuatro ORF dentro de él (fig. 1). El primero dirige la síntesis de los antígenos relacionados con el HBcAg y contiene dos señales de iniciación de la trascripción, que se corresponden con la síntesis del HBeAg y del propio HBcAg y definen dos regiones: la pre-core (pre-C) y la core (C). El segundo presenta una única señal de iniciación, ocupa las tres cuartas partes del genoma y dirige la síntesis de la ADN polimerasa viral (región P). El tercero contiene las secuencias que codifican las tres glucoproteínas de superficie y presenta tres señales de iniciación de trascripción que definen las regiones pre-S1, pre-S2 y S. El cuarto está situado en el extremo 59, es el más pequeño y consta de una única señal de iniciación y una única región, que se conoce como región X. Esta región codifica una proteína no estructural conocida como antígeno x (HBxAg) que es un potente transactivador de la trascripción, capaz de potenciar también la expresión de genes celulares y quizás implicado en la inducción de tumores. En el genoma existen, además, dos secuencias enhancer denominadas ENI y ENII, dos secuencias de repeticiones directas de 11 bases llamadas DR1 y DR2 situadas en el extremo 59 de ambas hebras, una señal de poliadenilación y un probable sitio de respuesta a glucocorticoides (GRE).
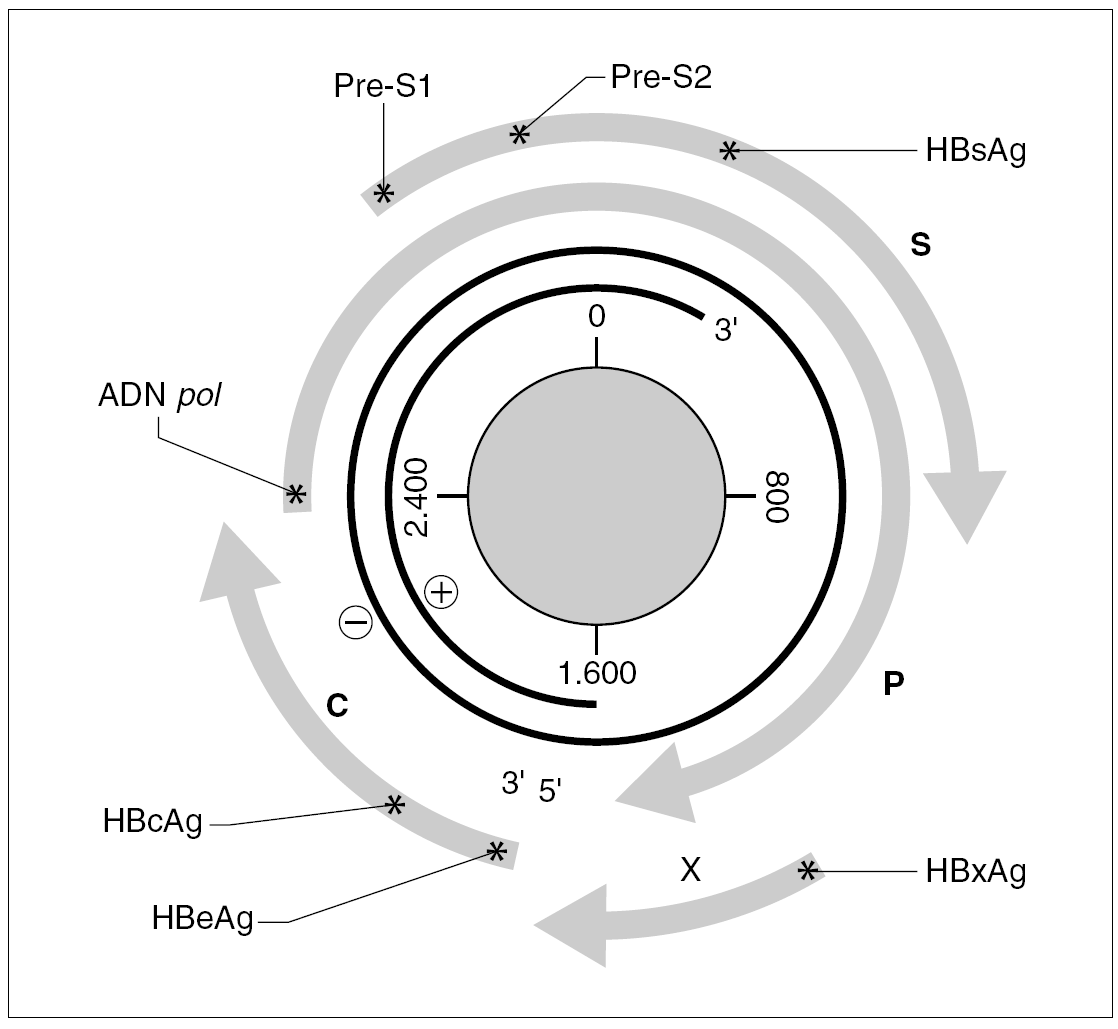
Figura 1. Organización del genoma del VHB y productos de su expresión. Los asteriscos indican la presencia de señales de iniciación de transcripción dentro de cada uno de los ORF (C, P, S y X). La transcripción a partir de cada señal origina un mensajero que se traduce en la proteína que se especifica en cada caso.
La adsorción del VHB a los hepatocitos se produce merced a la interacción entre las glucoproteínas de superficie y algún receptor celular todavía desconocido. La penetración tiene lugar por fusión de membranas. La replicación viral se desarrolla en dos fases, con el concurso de la ARN polimerasa II celular y de la ADN polimerasa viral 17. En la primera fase, el genoma entra en el núcleo, donde ocurre su conversión a un ADN circular covalentemente cerrado cuya cadena negativa es transcrita por la ARN polimerasa II celular para dar lugar a varios mensajeros. Uno de ellos produce el HBcAg y, por iniciación interna, la ADN polimerasa viral. Además, este segundo mensajero servirá también como molde para la retrotranscripción, que se produce durante la segunda fase, tras la salida del molde al citoplasma. El acoplamiento de la ADN polimerasa viral al ARN molde induce, además, la incorporación de dímeros de HBcAg, con formación de cápsides incompletas. De manera simultánea, otros mensajeros dirigen la síntesis de las glucoproteínas de la envuelta, que dimerizan durante su paso por el retículo endoplásmico y se van agregando en oligómeros para producir partículas virales vacías que emergen en el pre-Golgi. En este estado, con el ADN viral parcialmente sintetizado, las nucleocápsides que lo contienen se asocian a las glucoproteínas de superficie ancladas en las membranas del retículo endoplásmico y, más tarde, brotan a su través hacia el interior, adquiriendo así la envuelta. La liberación de los viriones sucede por un mecanismo de excreción a través de las cisternas del retículo endoplásmico, lo que permite que se complete el proceso sin producir roturas en la membrana celular y sin originar la muerte de la célula.
Al mismo tiempo que sucede la síntesis de partículas completas, el genoma viral dirige la síntesis de HBsAg en cantidades muy superiores a las necesarias para generar envueltas. Este exceso de HBsAg es internalizado por el retículo endoplásmico en forma de agregados, que son excretados por el mismo mecanismo al exterior de la célula. Por su parte, la expresión de la región pre-C origina moléculas de HBeAg que presentan en su extremo aminoterminal un péptido señal para excreción y son vertidas al medio extracelular tras la separación del péptido por proteólisis. Así, la célula infectada produce y libera, a un tiempo, viriones maduros, agregados de HBsAg y moléculas individuales de HBeAg, todo ello sin sufrir daños graves.
Durante todo este proceso, una parte de las proteínas estructurales del virus son procesadas por la maquinaria celular de reconocimiento de proteínas extrañas, y los péptidos que se generan son expuestos en la superficie celular en asociación con proteínas HLA de clase I. El HBcAg es el que genera los péptidos más inmunógenos y la respuesta inmunitaria celular se dirige, fundamentalmente, hacia epítopos contenidos en dicho antígeno.
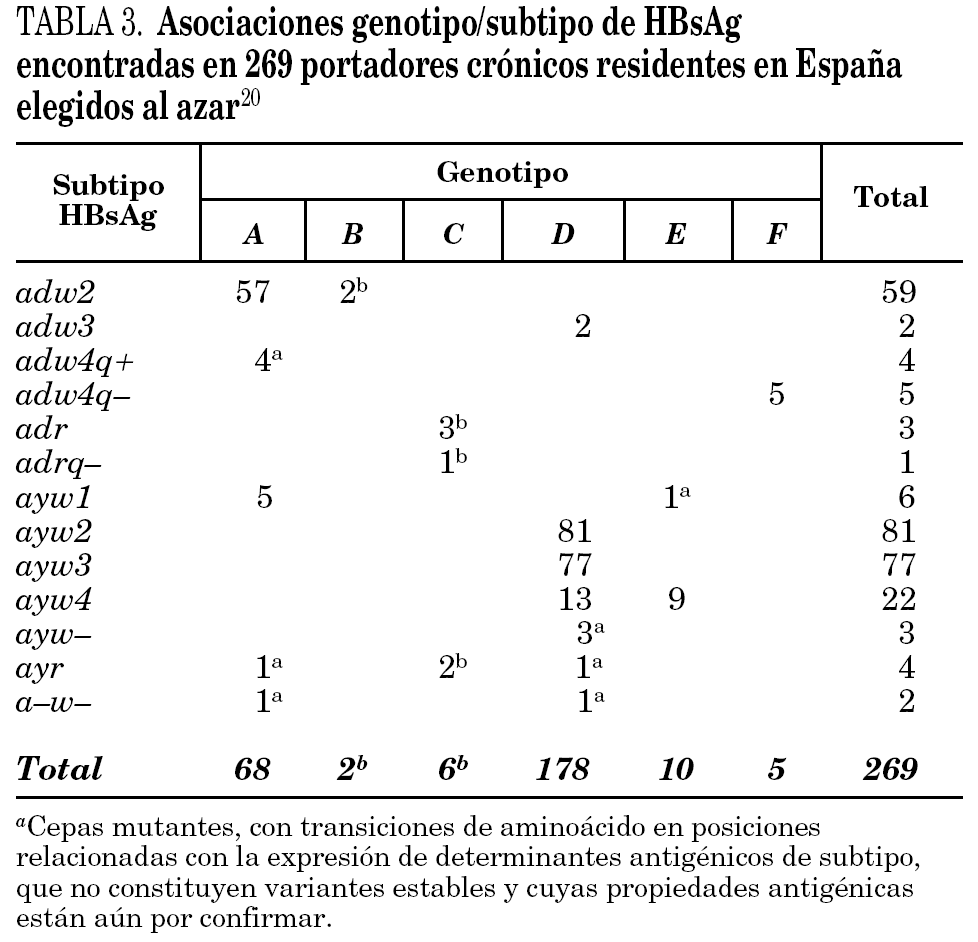
Se han identificado hasta la fecha ocho genotipos distintos (genotipos A-H) del VHB, así como algunos genosubtipos dentro de los genotipos A (A1-3), B (B1-3), C (C1-4), D (D1-4) y F (F1-2) 18,19. Los genotipos A y D son ubicuos, pero los restantes exhiben restricciones geográficas más o menos acusadas. Combinando el genotipo con las características inmunológicas del HBsAg (subtipos antigénicos), se diferencian hasta 20 grupos diferentes dentro del VHB, de entre los que destacan 11 grupos principales (tabla 2). Algunos de ellos son virtualmente exclusivos de determinadas regiones del planeta y mayoritarios en ellas. Así mismo, existe un grupo que es característico en los adictos a drogas por vía parenteral de cualquier lugar del mundo (D/ayw3)18. En España, los grupos D/ayw2, D/ayw3 y A/adw2 son los prevalentes 20 (tabla 3), con presencia minoritaria entre la población autóctona de los grupos exóticos A/ayw1, D/adw3, D/ayw4, E/ayw4 y F/adw4q-, este último importado de América hace ya siglos. Entre los inmigrantes procedentes del Extremo Oriente se detectan cepas de los genotipos B y C, no así aún entre la población autóctona. En la Europa occidental, el fenómeno de la inmigración está suponiendo la introducción de genotipos exóticos que podrían difundirse entre la población autóctona en el futuro, como ya sucedió en España con el genotipo F. Por tanto, cabe esperar que la epidemiología molecular del VHB sufra cambios durante los próximos años en esta región.

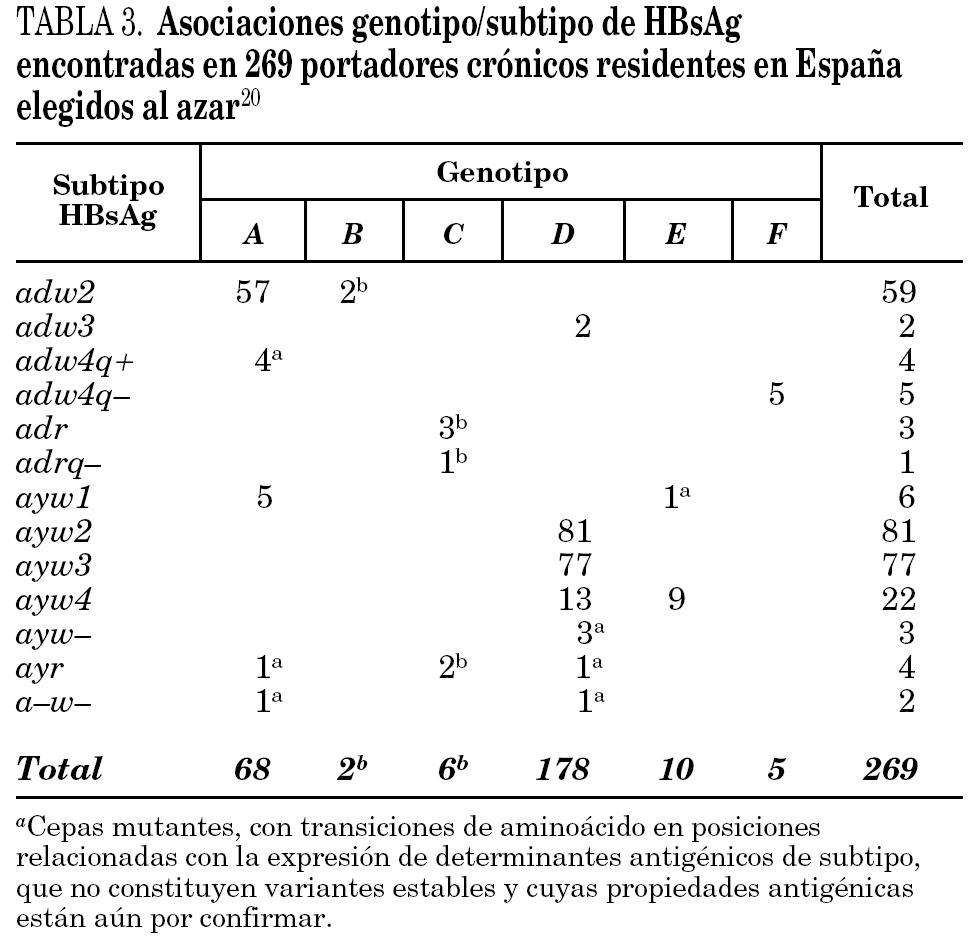
La presencia de un paso de retrotranscripción en la replicación del genoma del VHB hace que la frecuencia de mutación sea en este virus muy superior a la característica en los virus ADN, lo que permite al VHB evolucionar con mayor rapidez. Así, se han descrito variantes genéticas estables en todos los genotipos del virus, y algunas de ellas emergen como consecuencia de fenómenos de selección que, al estar condicionados por la respuesta inmunitaria o por la administración de productos terapéuticos, hacen que esas variantes sean de interés para la medicina y la salud pública 21 (tabla 4). El fracaso de la terapia con interferón a en pacientes con hepatitis crónica negativos para anti-HBe condujo al descubrimiento de las variantes pre-core defectivas, que son incapaces de sintetizar el HBeAg y que parecen emerger como consecuencia de la presión del sistema inmunitario del paciente durante una fase concreta de la infección crónica, prolongando la persistencia del virus 22. Posteriormente, el uso de compuestos inhibidores de la retrotranscriptasa (lamivudina, famciclovir, adefovir) como alternativa terapéutica al interferón ha mostrado la capacidad del VHB para generar mutantes resistentes que llegan a emerger durante el tratamiento y pueden condicionar su fracaso 23,24. Por último, las infecciones agudas en personas vacunadas frente al VHB con éxito, y el fracaso de la terapia con inmunoglobulina humana específica (HBIg) en niños nacidos de madres portadoras y en trasplantados hepáticos, han puesto de manifiesto la existencia de variantes capaces de escapar a la acción de los anticuerpos neutralizantes 25. Estas variantes presentan alteraciones en la secuencia de aminoácidos del HBsAg que afectan a la expresión del determinante antigénico a, que es común en todas las cepas salvajes de VHB conocidas y que garantiza la protección a largo plazo tras la infección primaria aguda resuelta, razón por la que se conocen como variantes a-defectivas. Algunas de estas variantes, en especial las que muestran ciertos cambios de aminoácido en la posición Gly145, pueden no ser detectadas correctamente por las pruebas de laboratorio que se utilizan para el diagnóstico de la infección, para la identificación de embarazadas portadoras y para el cribado de las donaciones de sangre y plasma, lo que viene a incrementar su interés sanitario 26.

Virus de la hepatitis D (agente delta)
Con un ARN monocatenario circular de 1,7 kDa y un único antígeno propio (el HDAg), el genoma del VHD no se parece al de ningún virus animal conocido y sólo se asemeja al de ciertos viroides y virusoides vegetales 27. En consecuencia, se cree que el VHD evolucionó a partir de un viroide vegetal que logró transgredir la barrera entre reinos y se adaptó a la especie humana. El VHD es un virus defectivo que no puede infectar sus células diana sin la ayuda del VHB, que contribuye a su ciclo vital aportando, al menos, las moléculas de HBsAg que este agente necesita para envolver sus cápsides. Así, el virión del VHD es una partícula quimérica que incorpora materiales procedentes de la expresión de dos genomas distintos.
El virión del VHD es una partícula esférica envuelta de 36 nm de diámetro. La cápside está formada por dos fosfoproteínas (p24 y p27) que sólo difieren entre sí en los últimos 19 aminoácidos. La más grande interactúa con el HBsAg y ancla la envuelta, que está constituida por este último. El genoma consiste en un ARN monocatenario y circular, covalentemente cerrado, de polaridad positiva, que adopta una compleja estructura secundaria superenrollada merced a la existencia de zonas de complementariedad interna que establecen apareamientos en el 70% de la secuencia. Este genoma se organiza en cinco ORF, de los que sólo el más largo codifica proteína. Al igual que sucede con los viroides y virusoides vegetales, el genoma del VHD es una ribozima, es decir, un ARN autocatalítico que es capaz de dirigir su propia replicación sin la participación de ninguna proteína funcional 28. Dicha replicación cursa mediante un mecanismo de rueda giratoria y a través de un ADN circular intermediario que se genera por trascripción inversa. El ciclo replicativo es citolítico.
Las cepas del VHD se clasifican en tres genotipos (genotipos I-III) 29. El genotipo I se halla ampliamente difundido en el mundo, y ocupa las mismas regiones en las que los genotipos A, D y E del VHB, originarios de África y Asia Central y Occidental, son prevalentes. Por su parte, el genotipo II solapa con los genotipos B y C del VHB, y se restringe al Extremo Oriente y el Sureste Asiático. Por último, el genotipo III es característico de las poblaciones indígenas de América y se solapa en su distribución con los genotipos F y H del VHB. Estos patrones coincidentes en diversificación y distribución indican una relación antigua entre ambos agentes, que habrían evolucionado juntos en cada una de esas regiones.
La infección por VHD puede suceder al unísono con la infección primaria aguda por VHB (coinfección) o con posterioridad a aquélla, siempre que el receptor sea portador crónico de VHB (sobreinfección). En el primer caso, la expresión del HDAg en los hepatocitos infectados estimula una respuesta inmunitaria vigorosa que da al traste con la estrategia de persistencia del VHB y conduce a la eliminación de ambos agentes, si bien causa con mayor frecuencia la aparición de cuadros graves de hepatitis aguda fulminante. Por el contrario, la sobreinfección del portador crónico conduce invariablemente a la persistencia de ambos y, con mucha frecuencia, acelera la evolución de la lesión hepática crónica, y desemboca rápidamente en cirrosis hepática.
Virus de la hepatitis C
El VHC es el único miembro oficialmente adscrito al género Hepacivirus dentro de la familia Flaviviridae. Su partícula es esférica y envuelta, con una nucleocápside icosaédrica formada por unidades repetitivas de una única proteína, conocida como antígeno core del VHC (HCcAg), y una envoltura lipoproteica que contiene dos glucoproteínas, una de transmembrana (E1) y otra formando espículas prominentes (E2).
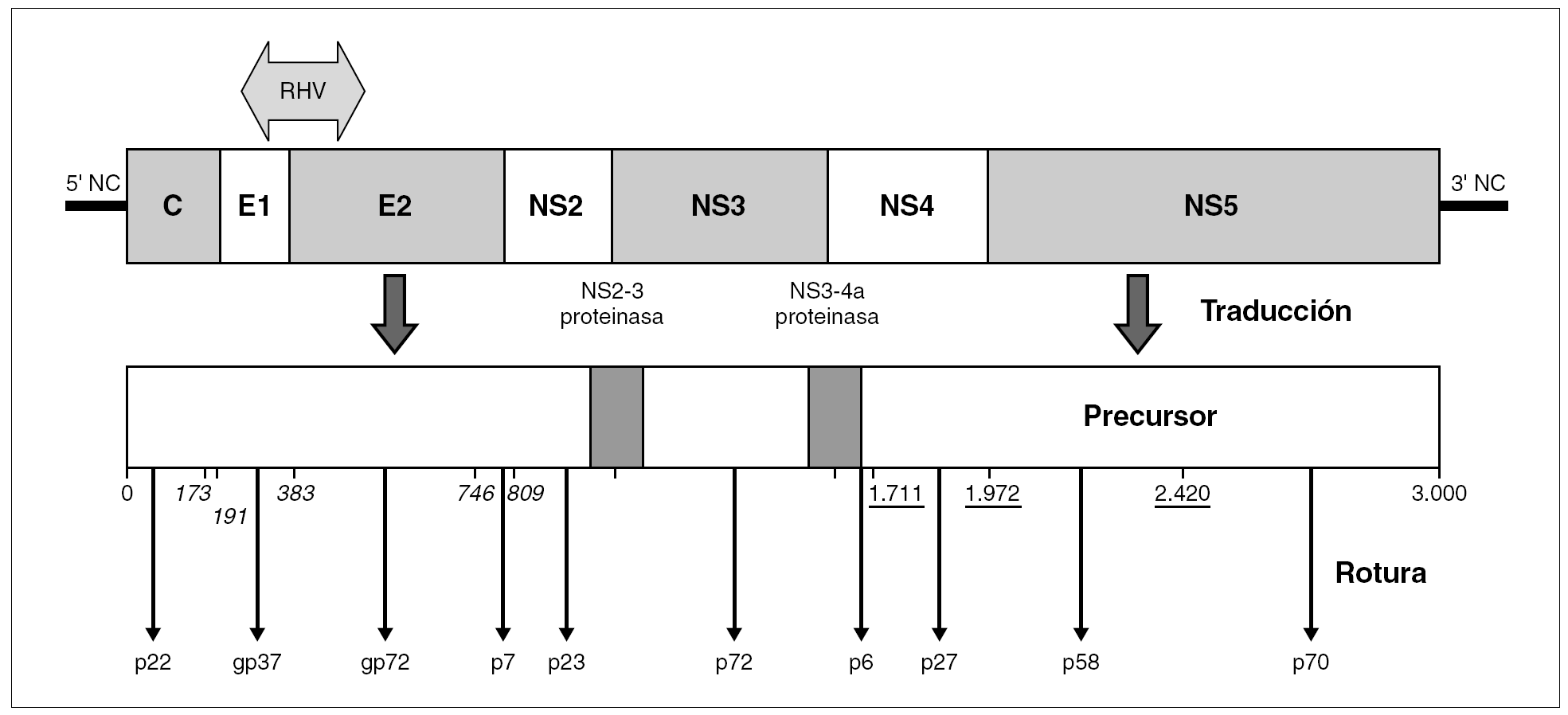
El genoma del VHC consiste en una molécula única y lineal de ARN monocatenario de, aproximadamente, 9,6 kb de tamaño y sentido positivo. En el interior del virión, la molécula se halla superenrollada y plegada en una estructura terciaria compleja. Por similitud con otros miembros de la familia y en virtud de sus productos de expresión y sus secuencias no codificantes, se distinguen en él nueve regiones (fig. 2): dos no codificantes situadas en los extremos (59-NC y 39-NC), tres estructurales en la mitad 59 (C, E1 y E2), y cuatro no estructurales en la mitad 39 (NS2-5) 30. Las regiones estructurales codifican el HCcAg, las glucoproteínas E1 y E2 y un pequeño polipéptido (p7) de función desconocida. Las no estructurales codifican proteínas funcionales, con actividades de metaloproteinasa o tiol-proteasa (NS2), serinproteasa, helicasa y nucleósido trifosfatasa (NS3), y ARN polimerasa dependiente de ARN (NS5b). Además, ciertas secuencias presentes en los productos de las regiones NS3 y NS4 colaboran en la actividad de las proteasas virales, que son las responsables del procesamiento de una parte de la poliproteína precursora.
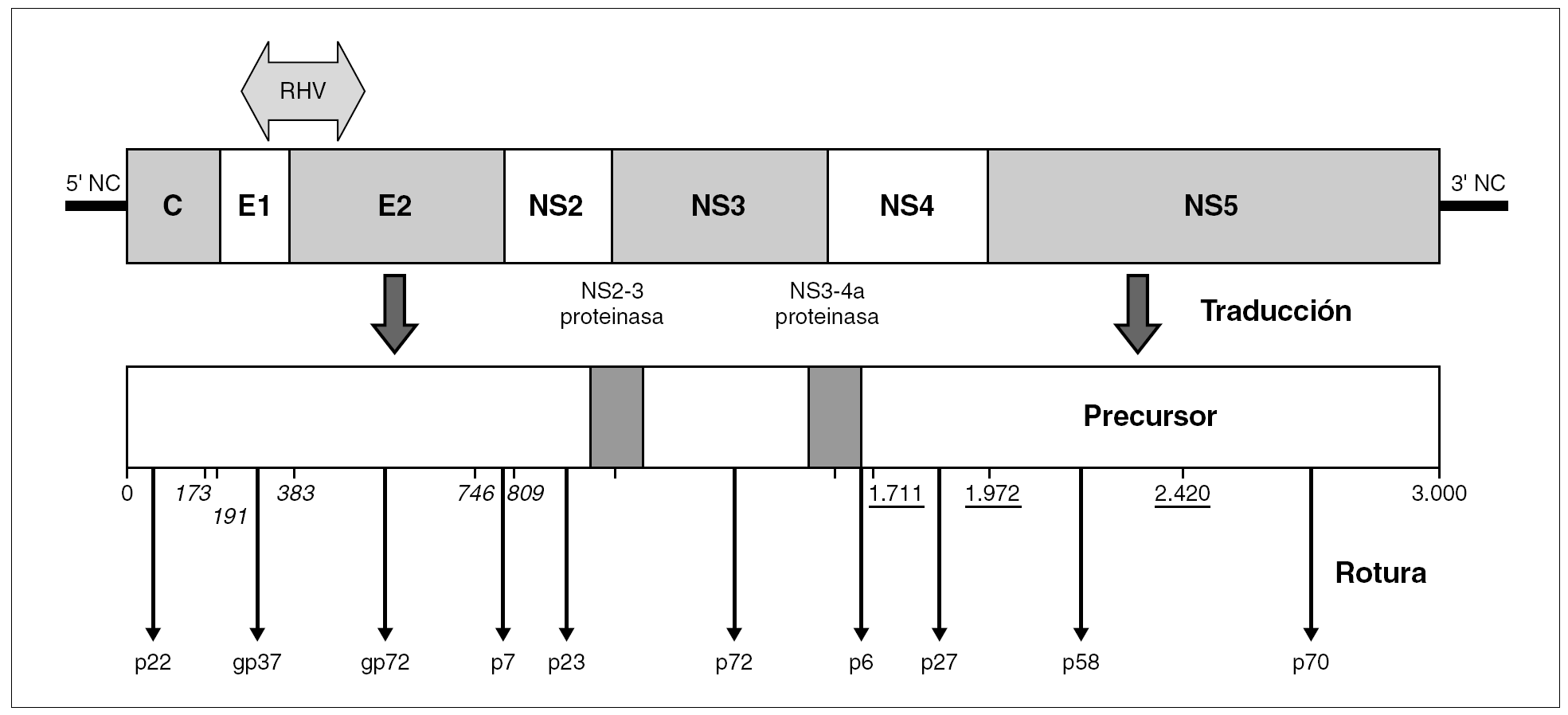
Figura 2. Organización del genoma del VHC y productos de su expresión. La popliproteína precursora única resultante de la traducción completa del ARN genómico es procesada por rotura mediante proteasas celulares (posiciones en cursiva) o mediante la serinproteasa viral (p27) (posiciones subrayadas). Las dos roturas restantes son realizadas por sendas actividades proteolíticas presentes en el precursor (NS2-3 proteinasa; NS3-4a proteasa), que involucran, en cada caso, aminoácidos que luego estarán presentes en dos proteínas virales diferentes. El genoma exhibe una región de alta variabilidad genética (RHV) situada en la unión de las regiones E1 y E2.
La adsorción de la partícula al hepatocito parece suceder a través de la interacción entre la glucoproteína E2 y el receptor celular de membrana conocido como CD81, aunque no puede descartarse que existan, también, otros receptores involucrados en el proceso 31. Posteriormente, las partículas penetran en la célula por un proceso de endocitosis en el que la fusión de las membranas estaría mediada por la glucoproteína E1. Una vez alcanzado el citoplasma, el ARN viral es traducido por los ribosomas merced a la actuación de secuencias de la región 59NC y de otras situadas en el extremo 59 de la región C. El resultado de la traducción es una poliproteína precursora de unos 3.000 aminoácidos cuyo procesamiento corre a cargo de proteinasas y señalasas celulares, de las actividades NS2-3 proteinasa y NS3-4a proteasa presentes en el propio precursor y de la serinproteasa viral. El procesamiento comienza por el extremo N-terminal con la actuación de las enzimas celulares, que liberan las proteínas estructurales (HCcAg, E1 y E2) y la proteína p7. Después, continúa en el orden preferente NS3/4a, NS5a/b, NS4a/b, NS4b/5a y se liberan las proteínas no estructurales. Los productos derivados de este proceso se asocian a las membranas del retículo endoplásmico formando el complejo replicativo típico de los virus ARN de sentido positivo. La ARN polimerasa viral es el componente clave para la replicación del genoma y se ha comprobado que puede iniciar dicha replicación de novo o mediante la extensión de un iniciador que es facilitado por el plegamiento y la hibridación interna de secuencias presentes en el extremo 39 del molde. La replicación sucede a través de un ARN intermediario de sentido negativo que sirve como molde para la síntesis de las nuevas copias del genoma. El ensamblaje de los viriones comenzaría con la interacción entre el HCcAg y el extremo 59 del ARN viral, induciendo el empaquetamiento de la cadena positiva e inhibiendo, así, la síntesis de proteínas. Al mismo tiempo, las moléculas del HCcAg interaccionarían entre sí a través del extremo N-terminal, formando la cápside en torno al genoma empaquetado. De forma paralela, las proteínas E1 y E2 se retendrían en la luz de las cisternas del retículo endoplásmico y la maduración de las partículas se produciría al brotar las nucleocápsides hacia el interior. Las partículas completas se exportan fuera de la célula por tránsito a través del aparato de Golgi, aunque lo cierto es que el VHC muestra una fuerte tendencia a permanecer asociado a componentes celulares.
Los análisis de las secuencias de cepas de VHC aisladas, con años de intervalo, de un mismo chimpancé infectado indican que la tasa de mutación en el genoma de este virus se sitúa en el intervalo de 1,44-1,92 * 103 sustituciones por base y año. Esta elevada tasa de mutación se debe a la también elevada tasa de error propia de las ARN polimerasas, que son enzimas que carecen de mecanismos de reparación de errores, aunque resulta significativamente más alta que la observada en otros virus ARN de sentido positivo (en torno a 104). Como es común en los virus ARN, los genomas del VHC presentes en el individuo infectado presentan una distribución en quasiespecies, es decir, forman un conjunto muy diverso de secuencias relacionadas pero diferentes entre sí, que se encuentran en distintas proporciones relativas. En el caso que nos ocupa, las especies moleculares que definen dicha distribución en cada momento cambian con el tiempo, y la máxima variación corresponde a las secuencias que codifican los extremos N-terminal y C-terminal, respectivamente, de las glucoproteínas E1 y E2, que definen la llamada región hipervariable (RHV) del genoma viral. Los epítopos involucrados en la neutralización por anticuerpos residen, precisamente, en los aminoácidos codificados en la RHV y, como veremos más adelante, estos hechos se consideran determinantes en la persistencia del VHC y en el mantenimiento de la infección crónica.
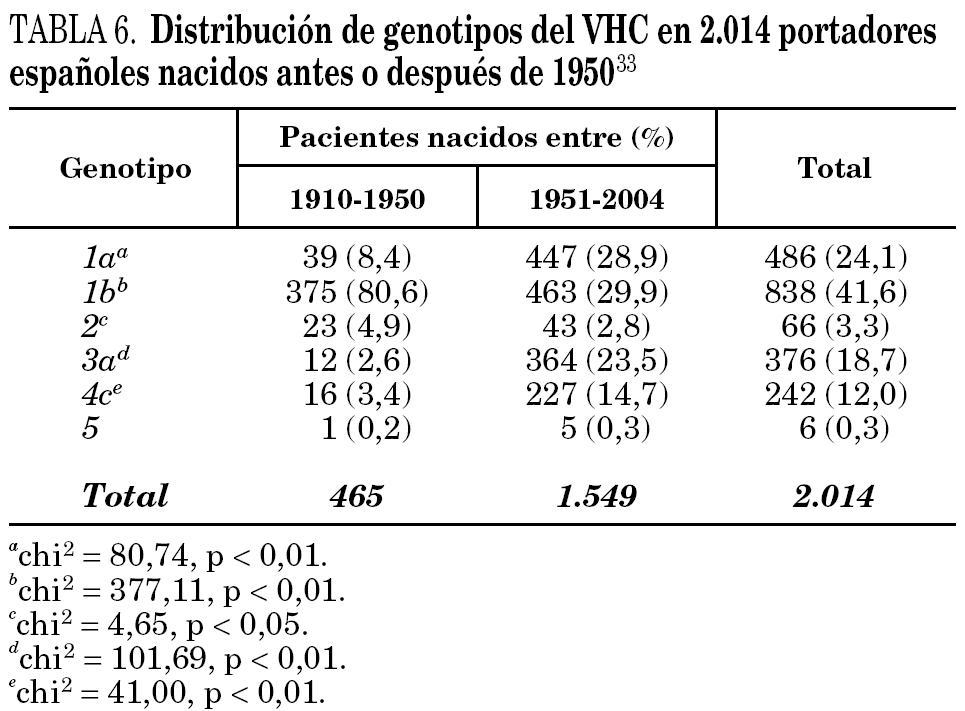
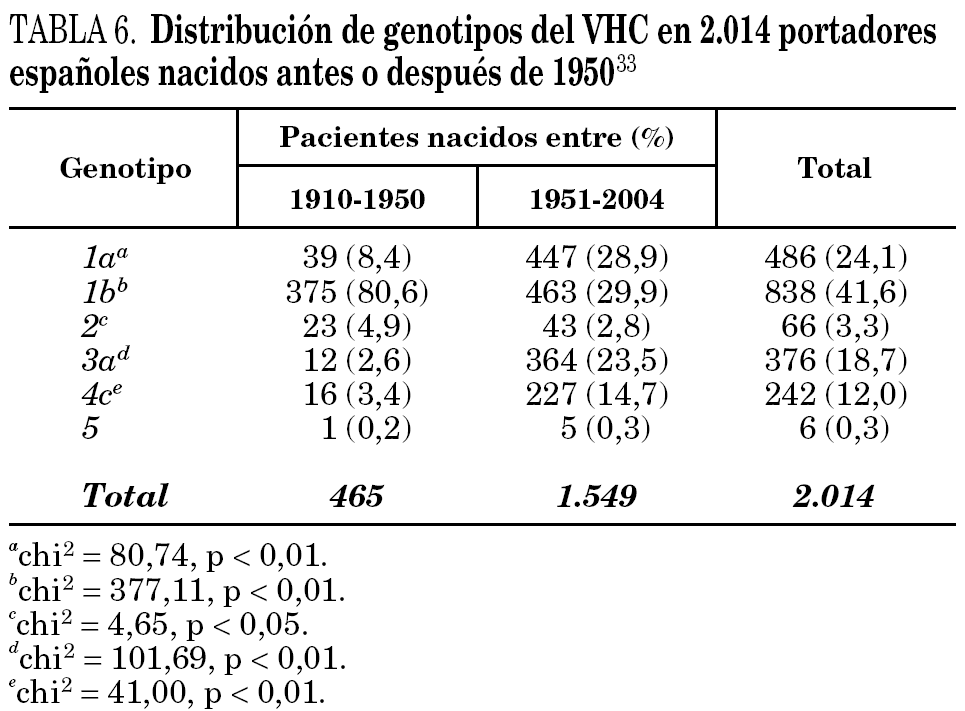
La comparación de las secuencias obtenidas de cepas de VHC procedentes de diferentes regiones del mundo revelan la existencia de seis ramas principales en los dendrogramas, que definen los seis genotipos aceptados hoy 32 (tabla 5). Con la única excepción del genotipo 5, pueden diferenciarse tres o más genosubtipos en cada uno de ellos. En esos dendrogramas, destaca la amplia diversificación que muestra el genotipo 6, que incluye en su agrupamiento dos genosubtipos y cuatro genotipos considerados inicialmente como independientes (genotipos 7, 8, 9 y 11), mientras que el descrito originalmente como genotipo 10 queda englobado en la rama correspondiente al 3. Los genosubtipos 1a y 1b se encuentran en todas las regiones del mundo, aunque el primero es prevalente en América y en el centro y norte de Europa, en tanto que el segundo lo es en el Extremo Oriente, en la Europa del Este y en la cuenca del Mediterráneo. El genotipo 3 es también ubicuo, pero presenta una especial prevalencia en el subcontinente Indio, en Australia y en las islas Británicas. El resto de los genotipos muestran, por el contrario, mayor restricción geográfica. Así, el genotipo 4 prevalece en el norte de África y no es raro en la Europa mediterránea, en tanto que el genotipo 5 es característico del sur de África y muy infrecuente fuera de esa región. En el África subsahariana, se detectan también los genotipos 1 y 2, incluyendo un genosubtipo (1c) que parece exclusivo de la región. Por último, el genotipo 6, entendido en sentido amplio, se halla muy localizado en el sureste de Asia, donde predomina claramente sobre el resto. Los datos obtenidos en España (tabla 6) sugieren la existencia de tres episodios principales de introducción de cepas del VHC 33. El primero habría supuesto la introducción de los genotipos 1b y 2 procedentes de África, resultando en un claro predominio del primero entre los portadores actuales de edad superior a los 50 años. El segundo correspondería a la introducción de los genotipos 1a y 3 en asociación al uso de drogas ilícitas de administración parenteral, y habría comenzado hace unos 25 años. El tercero, más reciente, estaría difundiendo cepas del genotipo 4, y se asociaría también a ese fenómeno. Estos dos últimos episodios coinciden en el tiempo con un aumento en la prevalencia del genotipo D del VHB en nuestro país y con la introducción del genotipo E y el grupo A/ayw1, ambos de origen africano, en la población española autóctona 20.

Mecanismos de persistencia viral en las hepatitis víricas crónicas
En las infecciones víricas agudas, los componentes humoral y celular de la respuesta inmunitaria específica logran la erradicación de la infección sobre la base de dos actuaciones principales: la neutralización por anticuerpos de los viriones infecciosos circulantes y la destrucción de las células infectadas por la acción de los linfocitos T citotóxicos específicos. Esto sucede en todos los pacientes que se infectan por los VHA y VHE y, virtualmente, en todos los que experimentan la coinfección VHB-VHD, así como en una gran mayoría de los que se infectan por VHB y en el 15-20% de los infectados por VHC. Sin embargo, el 5-10% de las infecciones posnatales por VHB, el 80-90% de las infecciones por VHC y prácticamente todas las sobreinfecciones por VHD en portadores de VHB desembocan en una infección persistente que, en ausencia de intervención terapéutica, se prolonga de por vida.
En el individuo inmunocompetente, la persistencia viral sólo es posible cuando el virus involucrado ha desarrollado alguna estrategia que le permite escapar a la vigilancia del sistema inmunitario. En la latencia, el virus se esconde en determinadas células y mantiene en silencio la práctica totalidad de las funciones de su genoma hasta entrar, en algún momento, en fase lítica. Cuando esto sucede, la recurrencia suele ser controlada rápidamente por el sistema inmunitario, lo que reestablece la situación anterior. En la infección persistente, existe síntesis de proteínas virales o, incluso, replicación viral completa, y esta situación convive con una respuesta inmunitaria activa que debería ser, en principio, capaz de erradicar la infección. En las infecciones persistentes por VHB y VHC, y en la mixta VHB-VHD, la persistencia cursa, o puede cursar, con replicación viral completa, lo que merece la consideración de infección crónica. Esta situación se mantiene sobre la base de dos mecanismos principales: la inducción de inmunotolerancia y la generación y selección de variantes de escape.
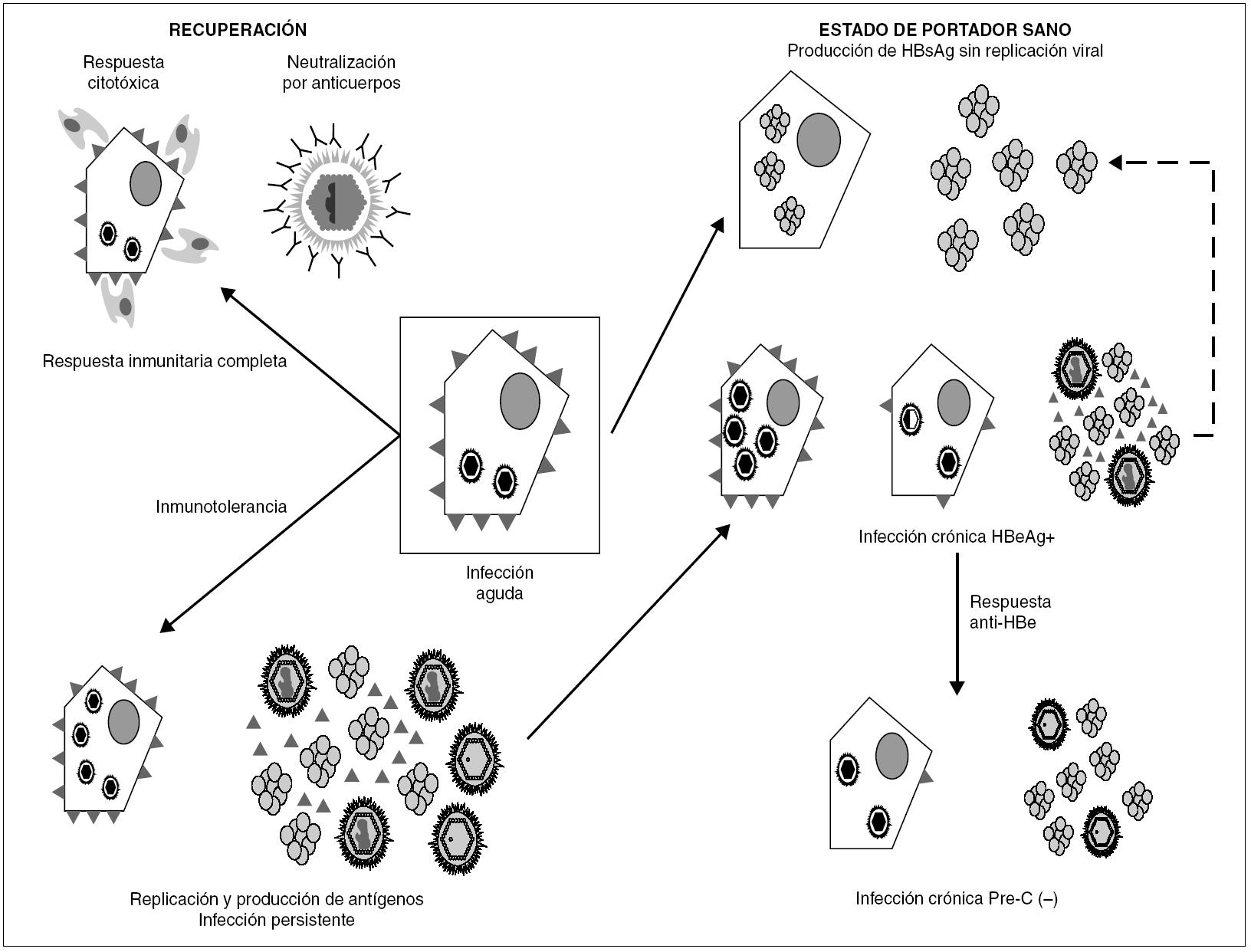
La persistencia del VHB en el portador crónico llega a involucrar ambos mecanismos (fig. 3). Inicialmente, los agregados de HBsAg y el HBeAg procedentes de los hepatocitos infectados bloquean, respectivamente, los sitios de unión de antígeno de los anticuerpos neutralizantes y los receptores de las células T (TCR), que reconocen los péptidos derivados del procesamiento del HBcAg que las moléculas HLA de clase I presentan en la superficie de esos hepatocitos. De esta forma, el VHB trata de bloquear los dos mecanismos principales de eliminación inmune, consiguiéndolo en el 5-10% de los casos de infección posnatal y en el 80-90% de las infecciones prenatales o perinatales. En esta fase temprana de la infección crónica, el paciente presenta concentraciones elevadas de HBeAg circulante, pero esa situación cambia en un cierto porcentaje de los portadores cuando, más tarde, se activa la respuesta de anticuerpos frente al HBeAg (anti-HBe) y se aclara totalmente el antígeno de la sangre. Este cambio debería, en principio, suponer la erradicación de la infección, toda vez que la respuesta citotóxica específica queda desbloqueada, y esto debiera originar la destrucción total de los hepatocitos infectados. Sin embargo, sucede que algunos pacientes que han entrado en esa nueva situación continúan replicando virus en el hígado durante años, y que el fenómeno se asocia a la presencia de variantes pre-core defectivas del VHB en su sangre. Se cree que la expresión de epítopos T en la superficie de los hepatocitos mayoritariamente infectados por dichas variantes es menor que en los infectados por variantes salvajes, y que, por tanto, los mutantes emergen como consecuencia de su capacidad de escapar al reconocimiento por la respuesta inmunitaria citotóxica, que constituiría el elemento ambiental de selección. El hecho de que este fenómeno de escape sea más frecuente en los portadores que viven en regiones geográficas en las que el genotipo D del VHB es más prevalente sugiere que la frecuencia de emergencia de estas variantes puede ser diferente en los distintos genotipos del VHB, y vendría a explicar por qué las hepatitis B crónicas anti-HBe positivas son especialmente frecuentes en la cuenca mediterránea.
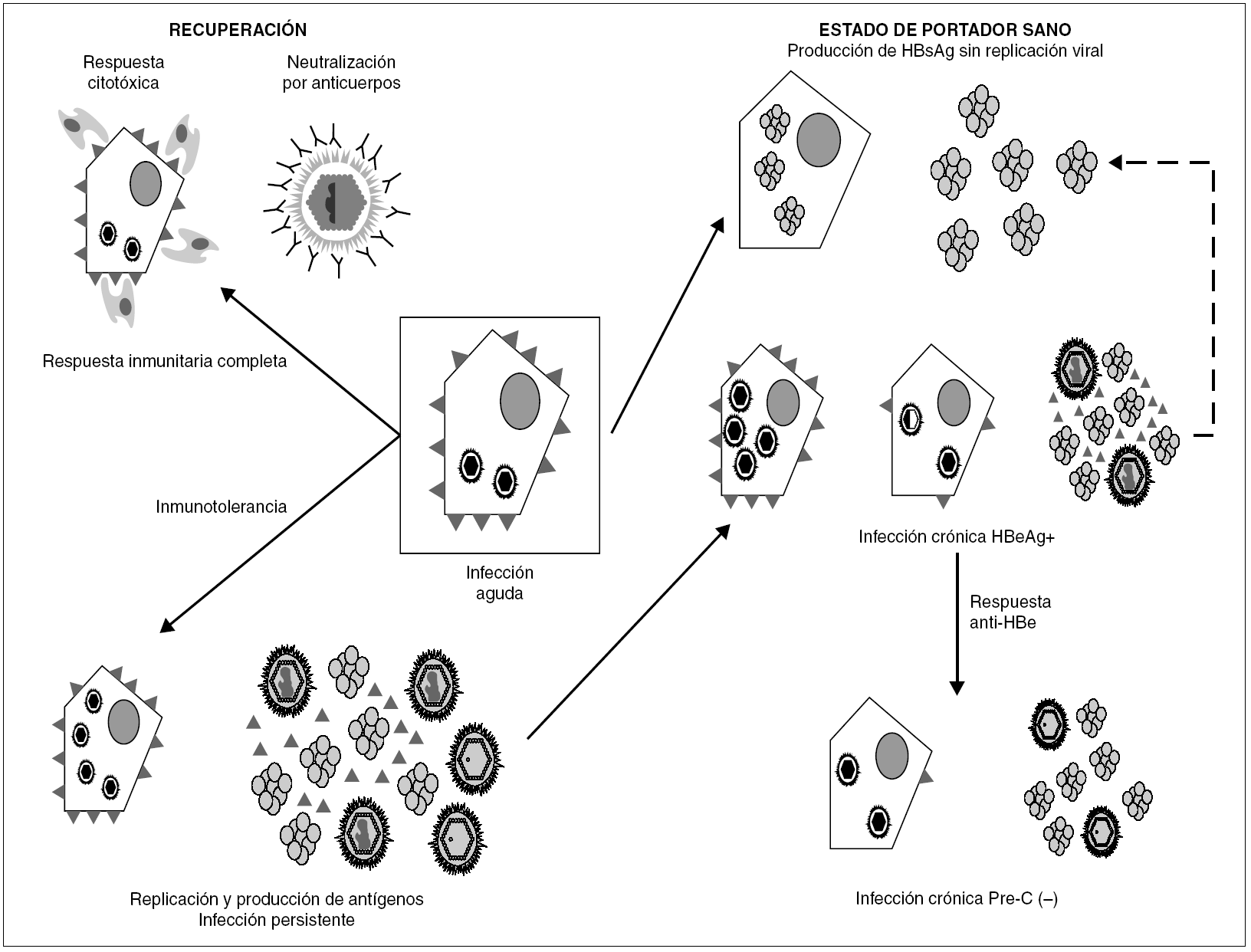
Figura 3. Esquema de los mecanismos de establecimiento y mantenimiento de la infección persistente por VHB. La persistencia se establece merced a la inhibición de los componentes humoral y celular de la respuesta inmunitaria, ejercida por los agregados de HBsAg y las moléculas individuales de HBeAg vertidas al torrente circulatorio. Una vez activada la respuesta de anti-HBe, aún puede mantenerse por selección de variantes pre-core defectivas, que parecen inducir una menor expresión de epítopos T en la superficie del hepatocito. En ocasiones, se llega a una situación de infección persistente con producción de HBsAg y sin replicación viral detectable ni expresión intracelular de HBcAg, situación que se conoce como estado de portador sano.
La sobreinfección por VHD introduce un cambio radical en cualquiera de las fases de la infección crónica en la que se encuentre el portador de VHB, ya que supone la aparición de nuevos epítopos T en la superficie de los hepatocitos infectados que inducen una respuesta inmunitaria citotóxica específica que el VHB no puede inhibir. Si el inóculo resulta en una rápida y masiva infección de hepatocitos, la propia citólisis que produce la replicación del VHD y la destrucción mediada por la respuesta inmunitaria se suman para producir un cuadro de hepatitis fulminante que conduce con frecuencia al fallo hepático agudo y a la muerte del paciente. En otro caso, la destrucción tisular progresa más despacio, pero conduce en poco tiempo a la cirrosis hepática. Por el contrario, en la coinfección aguda la respuesta citotóxica frente al VHD da al traste con la estrategia de persistencia del VHB, lo que resulta en la aparición de un cuadro autolimitado de hepatitis aguda y en la eliminación de ambos agentes.
Al margen de lo dicho hasta aquí, algunos datos sugieren la existencia de otros fenómenos, aún no bien caracterizados, que contribuirían a la persistencia del VHB. Así, no es raro documentar la reaparición de marcadores de replicación viral asociada a situaciones de inmunodepresión en pacientes con marcadores que indican infección por VHB pasada y resuelta 34, lo que sugiere la existencia de fenómenos de latencia y reactivación en esta infección. Por otro lado, el uso de técnicas ultrasensibles de amplificación genómica ha conducido al hallazgo de concentraciones muy bajas de ADN viral circulante en pacientes negativos para HBsAg e, incluso, positivos para anti-HBs, lo que parece indicar la existencia de infecciones crónicas en pacientes cuya respuesta inmunitaria específica se halla totalmente desbloqueada. Este fenómeno se conoce como hepatitis B oculta 35, y su significado biológico y clínico es aún desconocido.
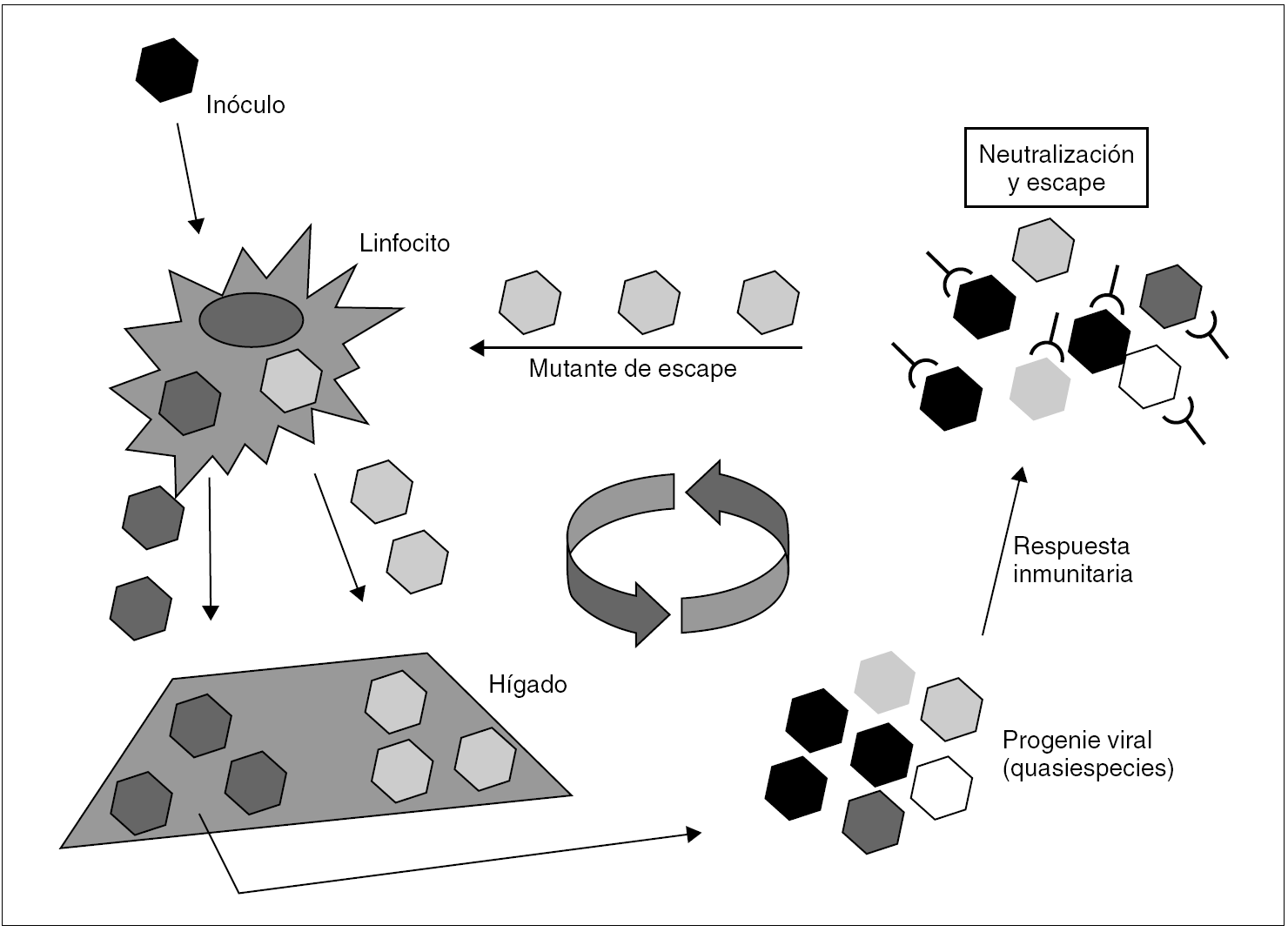
En el caso del VHC, no se conoce ningún mecanismo de inducción de inmunotolerancia en el huésped semejante al que ejercen el HBsAg y el HBeAg, aunque se ha sugerido que el HCcAg podría desempeñar un papel modulador de la respuesta inmunitaria que, quizá, contribuya a facilitar la persistencia del virus durante las fases tardías de la infección primaria 36. Está claro, en todo caso, que es la rápida emergencia y selección de mutantes de escape resistentes a la neutralización por anticuerpos lo que posibilita la persistencia del VHC y mantiene la infección crónica 37 (fig. 4). Así, cuando el inóculo es suficientemente diverso y tras la primera ronda de replicación en el hígado, el espectro antigénico de la progenie viral puede ser tan amplio como para desbordar la capacidad de la respuesta inmunitaria neutralizante, lo que resulta en el escape de una parte de la progenie, que llegará de nuevo al hígado. Si las sucesivas rondas de replicación que tienen lugar a continuación generan una diversidad antigénica suficiente en las sucesivas progenies virales, el fenómeno de escape se repite, y la diversidad se amplifica hasta alcanzar un grado que sobrepasa de forma definitiva la capacidad de respuesta del sistema inmunitario. Esta situación se alcanza en la gran mayoría de los casos y supone el definitivo asentamiento de la infección crónica. En consecuencia con todo lo anterior, la viremia y la elevación de las transaminasas en sangre son intermitentes durante los meses que siguen a la infección aguda. Más tarde, los valles de ambas curvas van siendo menos pronunciados y más espaciados. Transcurrido cierto tiempo, ambos parámetros se estabilizan. Ya que no parece que el VHC produzca una inhibición de la respuesta T citotóxica, ésta permanece activa y operante durante todo el proceso, lo que excluiría la existencia de portadores crónicos sanos de virus.
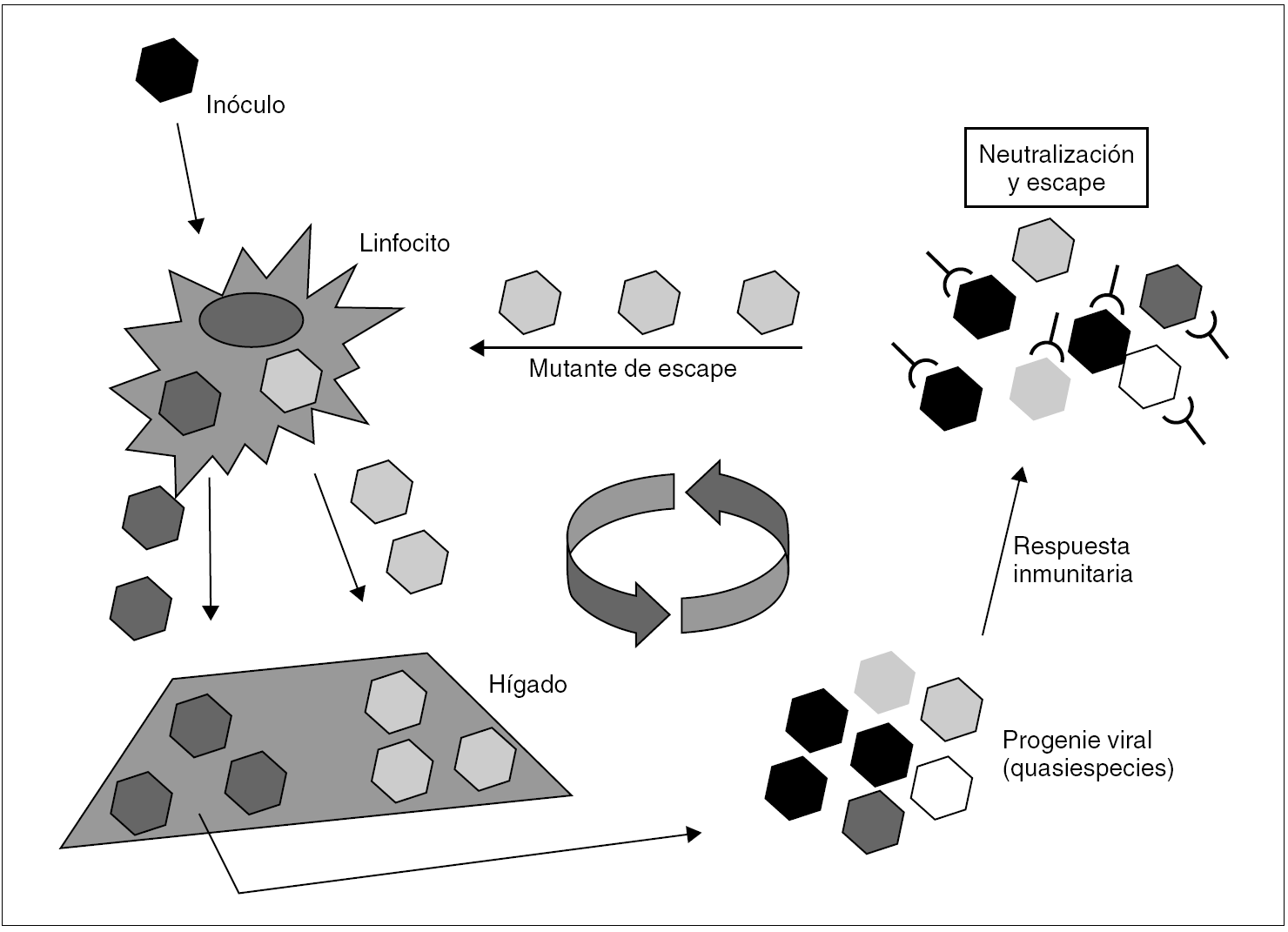
Figura 4. Esquema de los mecanismos de establecimiento y mantenimiento de la infección persistente por VHC. Un inóculo monoclonal imaginario de virus alcanza el hígado y replica en él para producir una progenie distribuida en una mezcla de variantes (distribución en quasiespecies). Si todas las variantes son reconocidas por la respuesta inmunitaria neutralizante (situación no prevista en el esquema), toda la progenie resulta neutralizada y la infección se controla y erradica. Si alguna o algunas variantes no lo son, se selecciona una población que alcanza de nuevo el hígado y replica en él. Esto último es lo que sucede en la gran mayoría de los casos, y es fácil comprender que resulta más probable cuanto más diverso es el inóculo inicial. La repetición cíclica de este proceso de mutación y selección de variantes de escape mantiene la infección crónica a largo plazo.
En conclusión, las hepatitis víricas forman un complejo conjunto de patologías originadas por agentes que son muy distintos en su biología y en su trascendencia en salud. Algunos son capaces de establecer infecciones crónicas a través de mecanismos cuyo conocimiento nos ha revelado detalles de enorme interés para el conocimiento general de los fenómenos adaptativos que gobiernan la evolución de la vida en sus formas más simples, además de ilustrarnos otros, más complejos, de enorme interés en el terreno específico de la infección y la enfermedad infecciosa.

NOTA
Los artículos publicados en la sección "Formación Médica Continuada" forman parte de grupos temáticos específicos (antibiograma, antimicrobianos, etc.). Una vez finalizada la publicación de cada tema, se irán presentando al Sistema Español de Acreditación de la Formación Médica Continuada (SEAFORMEC) para la obtención de créditos.
Una vez concedida la acreditación, esta se anunciará oportunamente en la Revista y se abrirá un período de inscripción gratuito para los socios de la SEIMC y suscriptores de la Revista, al cabo del cual se iniciará la evaluación, durante un mes, que se realizará a través de la web de Ediciones Doyma.
Correspondencia: Dr. J.M. Echevarría-Mayo.
Servicio de Microbiología Diagnóstica.
Centro Nacional de Microbiología.
Ctra. Majadahonda-Pozuelo, km 2. 28220 Majadahonda.
Madrid. España.
Correo electrónico: jmecheva@isciii.es
Manuscrito recibido el 6-7-2005; aceptado el 15-7-2005.

