




La aparición de múltiples variantes del SARS-CoV-2 durante la pandemia de COVID-19 es motivo de gran preocupación mundial. Hasta el momento, su análisis se ha centrado principalmente en la secuenciación de nueva generación. Sin embargo, esta técnica es costosa y requiere equipos sofisticados, largos tiempos de procesamiento y personal técnico altamente cualificado con experiencia en bioinformática. Para contribuir al análisis de variantes de interés y de preocupación, aumentar la capacidad diagnóstica y procesar muestras para realizar vigilancia genómica, proponemos una metodología rápida y fácil de aplicar, basada en la secuenciación Sanger de 3 fragmentos del gen que codifica para la proteína espiga.
MétodosSe secuenciaron 15 muestras positivas para SARS-CoV-2 con un valor de umbral de ciclo inferior a 25 por metodologías Sanger y secuenciación de nueva generación. Los datos obtenidos fueron analizados en las plataformas Nextstrain y PANGO Lineages.
ResultadosAmbas metodologías permitieron identificar las variantes de interés reportadas por la OMS. Se identificaron 2 muestras como alfa, 3 gamma, una delta, tres mu, una ómicron y 5 cepas cercanas al aislado inicial del virus Wuhan-Hu-1. Según el análisis in silico, también se pueden detectar mutaciones clave para identificar y clasificar otras variantes no evaluadas en el estudio.
ConclusiónLos diferentes linajes de interés y preocupación de SARS-CoV-2 se clasifican de forma rápida, ágil y fiable con la metodología de secuenciación de Sanger.
The emergence of multiple variants of SARS-CoV-2 during the COVID-19 pandemic is of great world concern. Until now, their analysis has mainly focused on next-generation sequencing. However, this technique is expensive and requires sophisticated equipment, long processing times, and highly qualified technical personnel with experience in bioinformatics. To contribute to the analysis of variants of interest and variants of concern, increase the diagnostic capacity, and process samples to carry out genomic surveillance, we propose a quick and easy methodology to apply, based on Sanger sequencing of 3 gene fragments that code for protein spike.
MethodsFifteen positive samples for SARS-CoV-2 with a cycle threshold below 25 were sequenced by Sanger and next-generation sequencing methodologies. The data obtained were analyzed on the Nextstrain and PANGO Lineages platforms.
ResultsBoth methodologies allowed the identification of the variants of interest reported by the WHO. Two samples were identified as Alpha, 3 Gamma, one Delta, 3 Mu, one Omicron, and 5 strains were close to the initial Wuhan-Hu-1 virus isolate. According to in silico analysis, key mutations can also be detected to identify and classify other variants not evaluated in the study.
ConclusionThe different SARS-CoV-2 lineages of interest and concern are classified quickly, agilely, and reliably with the Sanger sequencing methodology.
El síndrome respiratorio agudo severo coronavirus 2 (SARS-CoV-2) es un coronavirus que surgió en 2019 y es el responsable de la actual pandemia denominada enfermedad por coronavirus 2019 (COVID-19)1. Hasta la fecha, la COVID-19 se ha convertido en una amenaza para la salud pública mundial, causando aproximadamente 405 millones de infecciones y 5,8 millones de muertes en el mundo2.
El diagnóstico temprano es crucial para controlar la propagación de COVID-19. La detección molecular del ácido nucleico de SARS-CoV-2 mediante reacción en cadena de la polimerasa (PCR, por polymerase chain reaction) en tiempo real es considerada el estándar de oro. Sin embargo, puede afectarse por mutaciones del virus en las regiones 5′(UTR), ORF1ab, espiga (S), membrana y nucleocápside3,4, que favorecen el incremento en la tasa de falsos negativos5.
Las mutaciones en el genoma de SARS-CoV-2 generan cambios en las estructuras de las proteínas, afectando su virulencia, transmisibilidad y antigenicidad6,7. Por lo tanto, la vigilancia genómica del virus ha sido esencial para detectar tempranamente variantes de interés (VOI, por variant of interest) o de preocupación (VOC, por variant of concern). Todas las variantes presentan mutaciones en el gen de la proteína S4, por lo tanto, la mayoría de las estrategias terapéuticas y el desarrollo de vacunas se han centrado en este gen.
La detección de las diferentes mutaciones y la tipificación de las variantes se realiza por secuenciación de nueva generación (NGS, por next generation sequencing) del genoma completo de SARS-CoV-2. Las secuencias del virus son depositadas en la base de datos Global Initiative on Sharing All Influenza Data EpiCoV™, con el fin de compartir información genética de muestras positivas para SARS-CoV-28. Así mismo, existen plataformas que permiten realizar análisis genómicos y filogenéticos de las variantes, como Nextstrain9.
Aunque las técnicas de NGS son cruciales para la vigilancia molecular, exigen un alto grado de experiencia técnica, conocimientos en bioinformática y acceso a costosos equipos, que dificultan su uso masivo. Por lo anterior, se han diseñado otras metodologías, como secuenciación Sanger y PCR en tiempo real, como pruebas de tamizaje de variantes3,10.
En Colombia, las actividades de caracterización y vigilancia genómica de SARS-CoV-2 están centralizadas en el Instituto Nacional de Salud. Sin embargo, con el objetivo de contribuir a la vigilancia genómica, aumentar la capacidad diagnóstica y el flujo de trabajo simple, y mejorar el seguimiento de variantes en el menor tiempo posible, proponemos un método basado en secuenciación Sanger de 3 fragmentos que cubren parcialmente el gen que codifica para la proteína S para detectar VOI y VOC.
MétodosSelección de diana genómica y diseño de cebadoresSe utilizó la secuencia de referencia de SARS-CoV-2, GenBank: NC_045512, y el software NCBI-Primer BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/, https://www.ncbi.nlm.nih.gov/genbank/) para diseñar los cebadores. La calidad del diseño de los oligonucleótidos fue evaluada por OligoAnalyzer v3.1 (https://www.idtDNA.com/calc/analyzer). La especificidad de los cebadores se verificó mediante análisis de predicción in silico con la herramienta de búsqueda de alineación local básica en línea BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Simulación in silico de detección de variantes de interés y preocupaciónSe realizó la simulación de las posibles mutaciones en la secuencia de aminoácidos de la proteína S del GenBank. Se obtuvieron las secuencias de referencia de las VOC (alfa, beta, gamma, delta y ómicron) y VOI (lambda y mu). Se alinearon con el programa Sequencher 5.4.6 (Gene Codes Corporation) y la identificación de las variantes se realizó con el programa web Nextstrain-Nextclade Web 1.10.0, Nextclade CLI 1.7.0 (2021-12-09) (https://clades.nextstrain.org/).
Muestras de pacientes positivos para SARS-CoV-2Para el tamizaje de variantes de SARS-CoV-2, se seleccionaron 15 muestras clínicas de pacientes de Medellín (Colombia) con resultado positivo por PCR en tiempo real para COVID-19 entre el año 2020 y el 2021 con un valor de umbral de ciclo menor de 25. Las muestras fueron procesadas previamente mediante el protocolo de Charité-Berlín modificado, estandarizado y validado en el Laboratorio Integrado de Medicina Especializada11. Las muestras se tomaron siguiendo los lineamientos del Ministerio de Salud y Protección Social12.
Extracción de ARN y síntesis de ADNcEl ARN se obtuvo utilizando sistemas de extracción automatizada con tecnología de magnetos, KingFisher™ Duo Prime System (Thermo Fisher Scientific Inc., EE. UU.) con el kit MagMAX™ Viral/Pathogen Nucleic Isolation (Thermo Fisher Scientific Inc., EE. UU.). Igualmente, se realizaron extracciones manuales de algunas muestras con el Quick-RNA Viral Kit (Zymo Research, EE. UU.). El ARN total se convirtió en ADNc utilizando el kit TruScript™ First Strand cDNA Synthesis (Norgen,Thorold, Canadá), siguiendo las recomendaciones del fabricante.
PCR convencional para la detección de fragmentos de SARS-CoV-2Se realizaron 3 PCR convencionales independientes. Las condiciones de reacción fueron: 2μl Taq DNA Polymerase PCR Buffer 10X (Invitrogen, EE. UU.), 1,2μl MgCl2 50mM (ThermoFisher Scientific Inc., EE. UU.), 2μl dNTP Mix 10mM (Thermo Fisher Scientific Inc., EE. UU.), 0,3μl cebadores sentido y antisentido 20μM, 0,16μl Platinum Taq DNA Polymerase (Invitrogen, EE. UU.) y 2μl de ADNc para un volumen final de 20μl por muestra.
Las condiciones de termociclado fueron: 94°C×5min, seguido de 35 ciclos a 94°C×30s, 60°C×45s (excepto fragmento 3, 61°C×45s) y 72°C×3min. Finalmente, un ciclo a 72°C×7min. Se utilizó el termociclador ProFlex™ 3×32-well PCR System (Applied Biosystems™, EE. UU.) y los productos se visualizaron en un gel de agarosa al 2%.
Secuenciación por Sanger y análisis de variantesLos productos de PCR fueron cuantificados, diluidos a 40ng/μl y purificados con ExoSAP-IT™ for PCR Product Clean-Up (Applied Biosystems, Lithuania). Posteriormente, se secuenciaron en ambas direcciones utilizando el kit BigDye® Terminator v3.1 Cycle Sequencing (Applied Biosystems/Hitachi High Technologies, Japan)) y se purificaron con el kit BigDye® XTerminator™ Purification Kit. Finalmente, se realizó la electroforesis capilar en el equipo Genetic Analyzer 3500 (Applied Biosystems, Lithuania). Las secuencias consenso se generaron usando Sequencher 5.4.6 (Gene Codes Corporation, EE. UU.).
Para confirmar los resultados obtenidos de la secuenciación por Sanger, las muestras también fueron analizadas en la Unidad de Secuenciamiento de la Universidad de Antioquia, con el apoyo de los grupos PECET-Inmunovirología, de la misma universidad, mediante NGS usando los sistemas iSeq™ 100 (Illumina, EE.UU.), MiSeq™ (Illumina, EE.UU.) y Oxford Nanopore MinION (Oxford Nanopore Technologies, Reino Unido). La nomenclatura de variantes se realizó de acuerdo con el programa en línea Nextstrain-Nextclade Web 1.10.0, Nextclade CLI 1.7.0 (2021-12-09) y PANGO Lineages (https://cov-lineages.org/).
Análisis estadísticosLos datos de las mutaciones fueron tabulados y se analizaron utilizando el programa GraphPad Prism v7.04. La concordancia de la determinación de variantes entre la secuenciación por Sanger y por NGS se calculó mediante el índice kappa, considerando NGS como el estándar de oro y definiendo 2 posibles categorías (presente o ausente) en relación con las mutaciones o cambios evidenciados en la proteína S.
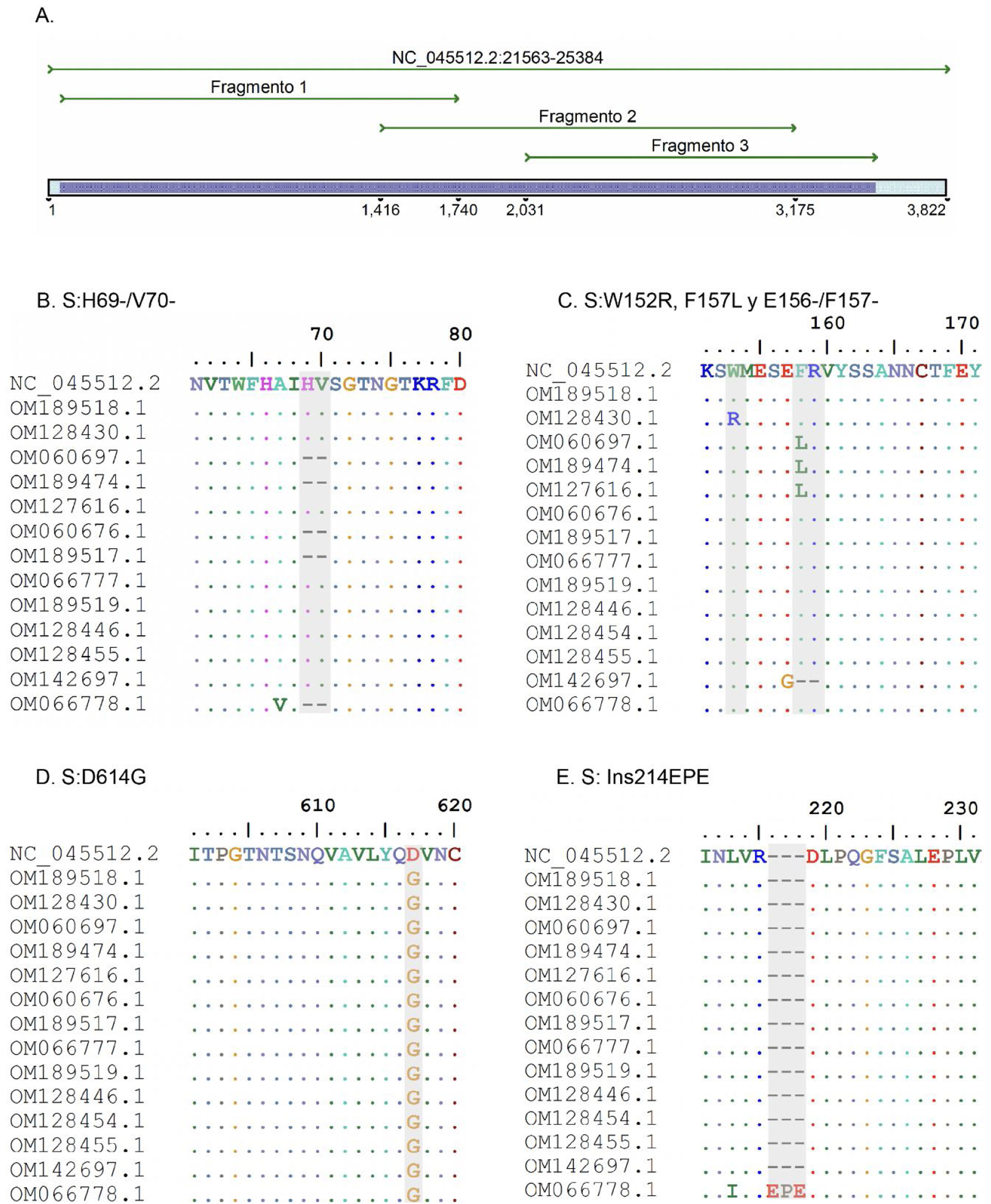
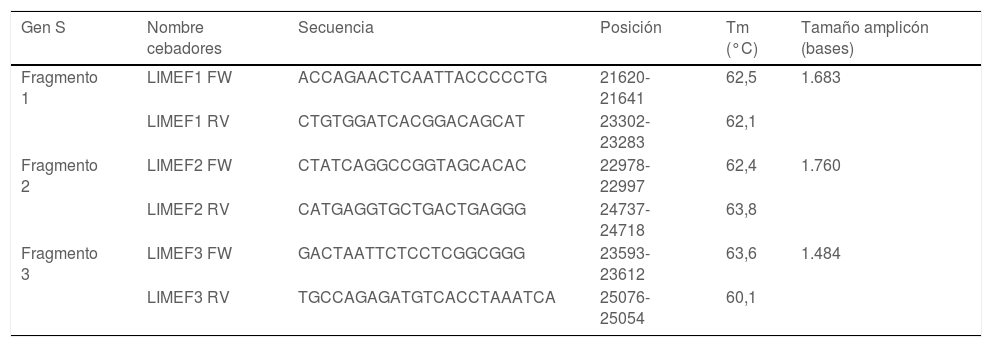
ResultadosSe diseñaron 3 pares de cebadores que inician en la posición 21.620 y terminan en la posición 25.076 de la secuencia de referencia NC_045512 del gen S del virus SARS-CoV-2, los cuales se listan en la tabla 1. Los amplicones generados se superponen entre sí y generan un producto de 3.434pb con una cobertura teórica del 89,9% del gen S (3.822pb), como también se aprecia en la figura 1.
Lista de cebadores diseñados
| Gen S | Nombre cebadores | Secuencia | Posición | Tm (°C) | Tamaño amplicón (bases) |
|---|---|---|---|---|---|
| Fragmento 1 | LIMEF1 FW | ACCAGAACTCAATTACCCCCTG | 21620-21641 | 62,5 | 1.683 |
| LIMEF1 RV | CTGTGGATCACGGACAGCAT | 23302-23283 | 62,1 | ||
| Fragmento 2 | LIMEF2 FW | CTATCAGGCCGGTAGCACAC | 22978-22997 | 62,4 | 1.760 |
| LIMEF2 RV | CATGAGGTGCTGACTGAGGG | 24737-24718 | 63,8 | ||
| Fragmento 3 | LIMEF3 FW | GACTAATTCTCCTCGGCGGG | 23593-23612 | 63,6 | 1.484 |
| LIMEF3 RV | TGCCAGAGATGTCACCTAAATCA | 25076-25054 | 60,1 |
Tm: temperatura de fusión (melting).
.")
A. Simulación in silico de la cobertura del gen S por los cebadores LIME. Los productos generados cubren la mayoría de las mutaciones y deleciones asociadas con VOI y VOC. B. Deleción H69/V70 frecuente en la variante alfa. C. Mutaciones puntuales y deleciones entre los aminoácidos 148, 150 y 160 de la proteína S; se destaca la deleción E156/F157 identificada en la variante delta. D. Mutación D614G presente en todas las muestras evaluadas. E. Inserción 214EPE característica de la variante ómicron 21K (BA.1).
Según el análisis in silico, las mutaciones presentes en las variantes reportadas hasta la actualidad no interfieren en el rendimiento y amplificación de los fragmentos. No obstante, se identificó un cambio de un solo nucleótido en el cebador LIMEF3 FW que amplifica el fragmento 3.
Análisis in silico de variantes de interés y preocupaciónSe realizó análisis in silico de las VOI y VOC reportadas hasta la actualidad en el Tracking SARS-CoV-2 variants de la Organización Mundial de la Salud. Teóricamente, con la metodología de Sanger se pueden detectar las mutaciones específicas S:G75V, S:T76I, S:F490S y S:T859N, y 7 deleciones de la variante 21G (lambda) que corresponden al linaje C.37. En el caso de la variante mu, es posible detectar las mutaciones relevantes S:R346K y S:Y144S, al igual que las 7 mutaciones también reportadas en las otras VOI y VOC.
Para las VOC, en alfa, los resultados mediante Sanger identificaron las mutaciones específicas S:A570D, S:T716I y S:S982A. Sin embargo, no se podría detectar la mutación ubicada en el aminoácido 1118 (S:D1118H). En el caso de la variante beta, se pueden detectar las mutaciones S:D80A y S:D215G y las deleciones S:L241-, S:L242- y S:A243-, y en gamma, se pueden detectar las variantes S:D138Y, S:R190S y S:T1027I. Para la variante delta, se podrían detectar las mutaciones compartidas por el clado 21A y 21J (S:R158G, E156- y S: F157-), pero no se podría diferenciar el subclado 21A de 21J debido a la presencia de mutaciones en los genes de la nucleocápside y ORF1a-b, que no son analizadas por la metodología Sanger. Finalmente, en el caso de la variante ómicron se encontraron 2 perfiles mutacionales cercanos entre sí, clados 21K y 21L. Para el clado 21K, se podrían detectar 8 mutaciones específicas, pero en el clado 21L solo se detectarían 7 mutaciones, lo que permitiría diferenciar entre ambos clados.
Análisis de variantes en muestras positivas para SARS-CoV-2Los amplicones obtenidos de las 3 PCR convencionales cubren experimentalmente 3.434 nucleótidos del gen S, correspondiente a 1.144 aminoácidos. Se obtuvieron secuencias Sanger de buena calidad en las 15 muestras clínicas. Se clasificaron 2 muestras alfa, 3 gamma, una delta, 3 mu, una ómicron y 5 cepas se relacionaron con el aislado inicial del virus Wuhan-Hu-1.
En la comparación en ciego de los resultados de la secuenciación por Sanger y NGS (estándar de oro), los resultados de clasificación de ambos métodos en las 15 muestras fueron concordantes (material suplementario, anexo 1).
Las muestras B1 (OM189518) y 5703 (OM128430) fueron clasificadas en el clado 19B, mientras que el resultado de NGS para ambas muestras las ubicó en 20A. Las mutaciones encontradas fueron S:D614G para la muestra B1, y en 5703 se encontraron las mutaciones W152R y D614G. Por su parte, las secuencias con los códigos 6041 (OM060697), 2403 (OM189474) y 12422 (OM127616) fueron diagnosticadas a principios del 2021 y clasificadas con el pangolineage B.1.625, clado 20A.
En cuanto a la identificación de las VOC, las muestras 11072 (OM060676) y BC08 (OM189517) fueron ubicadas en el clado 20I (alfa, V1). Las mutaciones constantes detectadas en estas muestras fueron 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I y S982A. Así mismo, 3 muestras se clasificaron como 20J (gamma, V3), correspondientes a los códigos 11896 (OM066777), BC22 (OM189519) y 28657 (OM128446). Las principales mutaciones detectadas fueron D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y y T1027I.
La muestra 36211 (OM142697) fue clasificada por Sanger como variante delta, pero fue ubicada en el clado 21A, mientras que por la metodología de NGS se ubicó en 21J. Ambos métodos de secuenciación muestran el mismo perfil de mutaciones en la proteína S. La muestra M1 (OM066778) fue categorizada como variante ómicron en el clado 21K, detectándose más de 36 mutaciones en la secuencia de aminoácidos de la proteína S.
También se logró la identificación de VOI. Las muestras 30535 (OM128454), 12932 (OM128455) y BC13 (OM301638) fueron clasificadas como mu y ubicadas en el clado 21H. Las mutaciones comunes detectadas fueron S:T95I, S:Y145N, S:R346K, S:E484K, S:N501Y, S:D614G, S:P681H y S:D950N.
DiscusiónLa comparación de los resultados de la secuenciación por Sanger y NGS de las muestras de COVID-19 evaluadas demostró que la metodología de Sanger logra la clasificación correcta de diferentes linajes de SARS-CoV-2, con resultados coherentes a los obtenidos por NGS (material suplementario, anexo 1).
El análisis estadístico con el índice kappa fue de 0,146 (IC95% −0,032 a 0,325), evidenciando un leve acuerdo entre métodos, lo cual probablemente está asociado a que ninguna de las metodologías detecta la totalidad de las mutaciones reportadas en la base de datos Tracking SARS-CoV-2 variants. Cabe resaltar que la metodología Sanger detecta las mutaciones claves para identificar el clado al que pertenece una cepa.
Aunque técnicamente no es posible amplificar y secuenciar por la metodología Sanger las 3.822 bases del gen S en una sola reacción13, en nuestro estudio proponemos la amplificación de 3 fragmentos que se solapan para lograr el 90% de cobertura del gen S. Lo anterior resulta eficiente en comparación con otras metodologías de secuenciación13,14, ya que acorta el tiempo de procesamiento, no requiere un número mínimo de muestras para el análisis, disminuye el número de reacciones necesarias para la obtención de resultados y reduce los costes asociados.
La metodología propuesta en este artículo requiere una adecuada toma, conservación y transporte de las muestras, que asegure un ARN íntegro con valores de umbral de ciclo menores de 25. Estos datos son similares a los recomendados en otras investigaciones13,15. El protocolo se probó con muestras extraídas por métodos automatizados de perlas magnéticas y extracciones con columnas con sílica, generando buen resultado, especialmente con la metodología de columnas.
Aunque no se encontraron todas las variantes de SARS-CoV-2 en las muestras clínicas, el análisis in silico de la proteína S demostró que esta metodología asegura la detección de los aminoácidos de interés S:K417, S:L452, S:S477, S:T478, S:E484, S:F490, S:S494 y S:N501. Estas mutaciones son de gran importancia debido a que la OMS y los CDC de EE. UU. las definieron como mutaciones clave presentes en VOI y VOC con fines de vigilancia genómica13.
La mutación S:D614G se encontró en todas las muestras analizadas, es llamada la mutación universal y se considera que todas las VOI y VOC derivan de ella16. S:D614G está relacionada con el aumento de la replicación viral en el tracto respiratorio superior e incrementa la eficiencia de la infección15. Esta ventaja adaptativa está relacionada con el aumento de los casos de COVID-19 en algunos países europeos a mitad de 2020 y su posterior dispersión en todo el mundo17,18.
La variante alfa está asociada con múltiples mutaciones en la proteína S, siendo la más destacada S:N501Y y la deleción 69-70. Esta última genera fallo de algunos ensayos de PCR en tiempo real5,19 y se encuentra en otras variantes, como 20A, 21K (ómicron) y 21L (ómicron).
Según los datos de covariantes, las mutaciones S:A570D, S:T716I, S:S982A y S:D1118H están asociadas con la variante alfa16. En este estudio no se pudo detectar la mutación S:D1118H por falta de cobertura en esa región, sin embargo, las mutaciones identificadas son suficientes para categorizar la variante.
Gamma está asociada con múltiples mutaciones; las específicas son S:N501Y, S:E484K, S:L18F, S:K417T y S:H655Y. Son compartidas por varias VOC, excepto la variante delta16. Nuestra metodología es capaz de detectar alrededor de 8 de las mutaciones características, incluidas 3 mutaciones específicas: S:D138Y, S:R190S y S:T1027I.
La variante delta, junto con la variante ómicron, son consideradas VOC y su rápida diseminación es la causante de los brotes desde mediados de 202120. La variante delta presenta 2 subclados; los resultados de Sanger y NGS analizados en Nextstrain generaron la clasificación 21A y 21J, respectivamente, para la muestra 36211. La discrepancia obtenida por los métodos de Sanger y NGS se debe a que el subclado 21J contiene todas las mutaciones del clado 20A con mutaciones adicionales en el gen N (N:G215C), al igual que en ORF1a (ORF1a:A1306S, ORF1a:V2930L, ORF1a:T3255I, ORF1a:T3646A) y ORF1b (ORF1b:A1918V, y ORF7b:T40 I)16,21, que no son analizadas en este estudio.
La variante ómicron surgió en noviembre de 2021; las primeras infecciones y datos de secuencia son predominantemente de Sudáfrica22. La identificación de esta variante es muy importante por ser la más frecuente actualmente, sus mutaciones están asociadas con una mayor transmisibilidad, escape inmunitario y mayor afinidad de unión a ACE223,24. Comparte 2 mutaciones con la VOC alfa, pero presenta alrededor de 26 mutaciones específicas16, las cuales son detectadas en su mayoría por el método de Sanger.
La VOI mu surgió a principios de 2021 y fue muy frecuente en Colombia16. Esta variante tiene mutaciones que también se encuentran en VOC y VOI, como S:T95I, S:E484K, S:N501Y, S:P681H y S:D95N. La mutación S:R346K ha sido considerada como específica de la variante mu, aunque esta mutación se presenta también en la variante ómicron.
De las variantes beta y lambda no fue posible obtener muestras para realizar el análisis experimental; los análisis in silico confirman que el método de secuenciación Sanger puede detectar las mutaciones características.
Nuestro grupo reconoce que el número de muestras analizadas es bajo pero permite demostrar que la metodología tiene la capacidad de categorizar experimentalmente VOC y VOI usando aplicaciones en línea de libre acceso, sin requerir la creación de algoritmos subjetivos. Para estudios posteriores recomendamos un mayor número de muestras y otras variantes no incluidas en el estudio.
Esta metodología supera algunas de las limitaciones de los métodos de PCR en tiempo real con sonda25, intercalante26 y alelos específicos27, donde es necesario el uso de cebadores y/o sondas específicas para evaluar cada mutación y con conocimiento previo de las mutaciones circulantes. Adicionalmente, algunas de estas PCR no son específicas para determinar variantes genéticamente relacionadas (subclados), no permiten evaluar la aparición de nuevas variantes y subestiman el número real de variantes circulantes.
Recientemente, se han publicado metodologías de secuenciación por Sanger que estudian la proteína S, e incluso ya están disponibles protocolos comerciales estandarizados28–30. Varios de esos estudios requieren un número mayor de cebadores dirigidos hacia dominios de interés, como RBD y RBM30, así como también analizan las mutaciones en otras regiones28, y en algunos casos hacen una secuenciación total de la proteína S. Todas esas metodologías anteriores implican realizar múltiples PCR29, lo cual aumenta el coste en comparación con la metodología que aquí proponemos.
Es importante resaltar que este enfoque no sustituye la tecnología de NGS y otros ensayos basados en PCR, más bien busca aumentar la capacidad de la red de vigilancia epidemiológica, al utilizarse como método de tamizaje de variantes, y en los casos en los que las NGS tengan poca cobertura o profundidad, puede utilizarse como estrategia para completar las secuencias.
En conclusión, esta metodología permite la detección de mutaciones en la proteína S de forma ágil y precisa, rápida y con infraestructura disponible en los laboratorios clínicos, de salud pública y de investigación en Colombia que sirven como apoyo a la red de diagnóstico y vigilancia genómica del virus SARS-CoV-2.
FinanciaciónEl presente trabajo fue financiado con recursos del sistema general de regalías de Colombia (BPIN 2020000100152) y de la Universidad de Antioquia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

