




La granulomatosis eosinofílica con poliangeítis (GEPA), previamente conocida como síndrome de Churg-Strauss (SCS), es una enfermedad multisistémica caracterizada por la presencia de asma, eosinofilia en sangre periférica y en los tejidos, formación de granulomas extravasculares y vasculitis de vasos de pequeño y mediano tamaño1. Es una enfermedad rara con una prevalencia de 10,7 a 13 casos por millón. El órgano más frecuentemente afectado es el pulmón. El tracto gastrointestinal (TGI) puede estar implicado en el 20% al 50% de los enfermos, pero la presentación clínica con sintomatología digestiva es infrecuente2.
Presentamos el caso de una paciente con GEPA que se manifiesta clínicamente como una colitis.
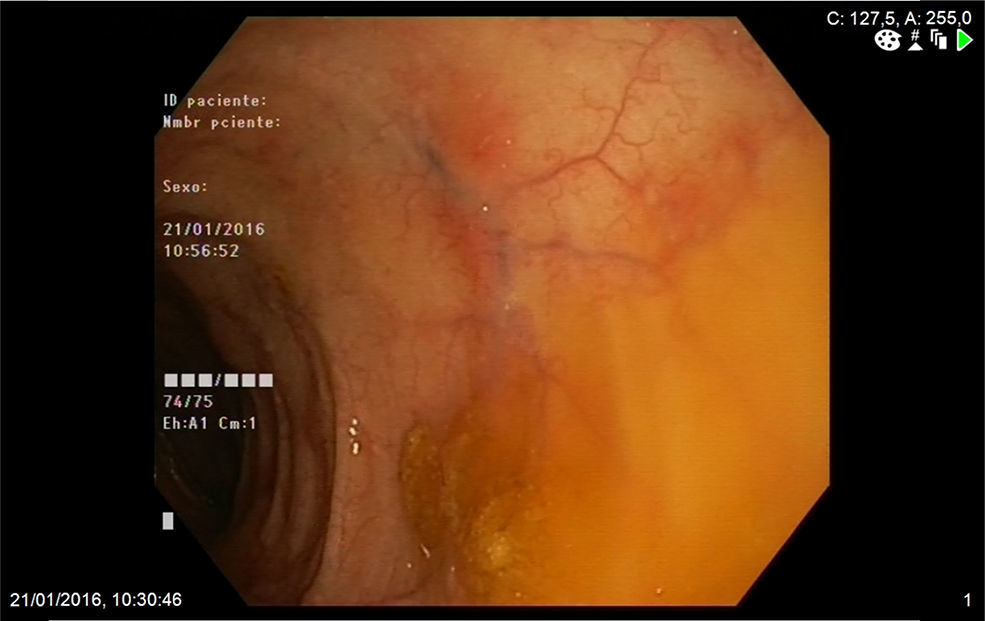
Mujer de 74 años que ingresa en nuestro servicio por clínica de 2 meses de evolución consistente en diarrea de 6 a 7 deposiciones al día con sangre y moco y dolor abdominal. En las 2 últimas semanas refería además tos con expectoración verdosa sin fiebre. Entre sus antecedentes personales destacaba una historia de asma de comienzo tardío, bronquiectasias, rinitis y sinusitis maxilar. En la exploración física estaba afebril y el único hallazgo reseñable era la presencia de crepitantes bibasales en la auscultación pulmonar. En la analítica realizada destacaba la presencia de leucocitosis, con 47% de eosinófilos, proteína C reactiva de 13,4mg/l (0,03-5) e IgE total de 271 kU/l (1,3-165). El frotis de sangre periférica solo demostró eosinofilia. El resto de estudios que incluyeron función renal y hepática, elemental de orina y niveles de vitamina B12 mostraron valores normales. Los anticuerpos antinucleares eran positivos a título 1:320 (patrón moteado) y los anticuerpos anticitoplasma de neutrófilo tipo mieloperoxidasa (ANCA-MPO) fueron negativos. Se realizó además determinación de parásitos en heces y un Mantoux, resultando ambos negativos. La radiografía de tórax demostró la presencia de infiltrados intersticiales en las bases. A continuación se completó el estudio con un ecocardiograma que mostró mínimo derrame pericárdico; una tomografía computarizada torácica que reveló afectación pulmonar bilateral, con áreas de consolidación subpleurales y áreas en vidrio deslustrado de predominio periférico y una colonoscopia en la que se apreciaba en el colon izquierdo áreas eritematosas parcheadas con abundante secreción mucoide que fueron biopsiadas (fig. 1). La biopsia de colon mostró una lámina propia congestiva con numerosos eosinófilos y una submucosa con un infiltrado inflamatorio de predominio eosinofílico de distribución perivascular (fig. 2A), así como oclusión de algunas luces vasculares y dudosos fenómenos de necrosis fibrinoide (fig. 2B) Ante todos estos hallazgos: asma, sinusitis, eosinofilia, infiltrados pulmonares y biopsia de colon con vasculitis y eosinofilia marcada se diagnosticó el paciente de GEPA y se inició tratamiento con prednisona a dosis de 1mg/kg/día. La paciente fue seguida en consultas con buena respuesta clínica y analítica, iniciándose descenso de esteroides a las 6 semanas e inicio de tratamiento con azatioprina a las 14 semanas.

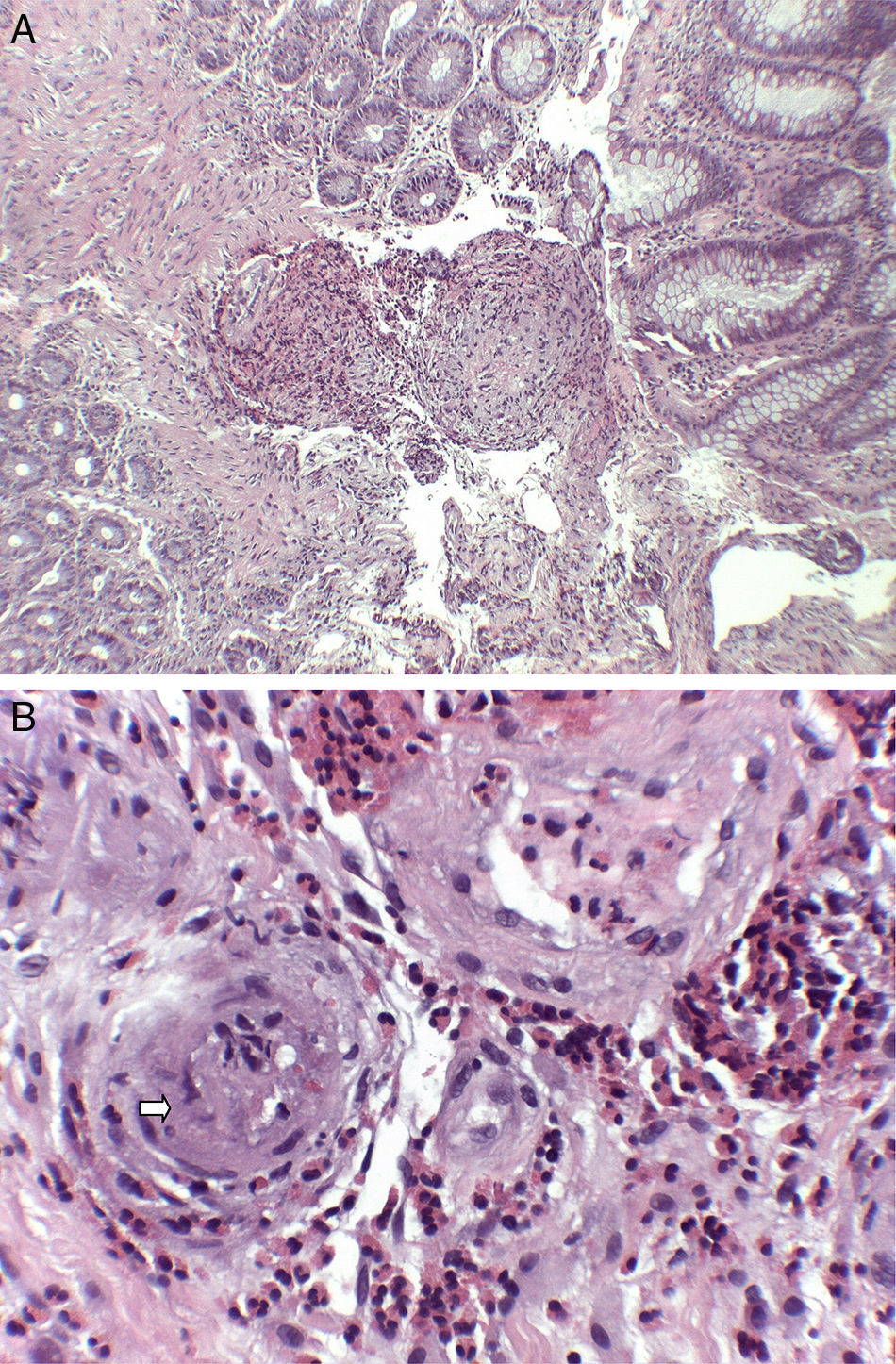
: A. Submucosa con infiltrado inflamatorio de predominio eosinofílico de distribución perivascular y marcado engrosamiento de las paredes vasculares (H-E×40). B. A mayor aumento se resalta la permeación de paredes vasculares por el infiltrado esosinofílico, con oclusión de algunas luces vasculares (flecha) (H-E×200).")
Hallazgos histológicos (tinción hematoxilina-eosina): A. Submucosa con infiltrado inflamatorio de predominio eosinofílico de distribución perivascular y marcado engrosamiento de las paredes vasculares (H-E×40). B. A mayor aumento se resalta la permeación de paredes vasculares por el infiltrado esosinofílico, con oclusión de algunas luces vasculares (flecha) (H-E×200).
La GEPA es una vasculitis sistémica que pertenece al grupo de vasculitis asociadas a ANCA. A pesar de esta clasificación solo se detectan los ANCA en un 30% a 40% de pacientes con GEPA. Sus manifestaciones clínicas se desarrollan en varias fases: una fase prodrómica que se manifiesta con síntomas de atopia, una fase eosinofílica caracterizada por eosinofilia periférica y tisular y una fase vasculítica en la que aparecen datos de vasculitis afectando a múltiples órganos como la piel, el sistema nervioso, el corazón, el riñón y el TGI3. El American College of Rheumatology ha propuesto 6 criterios para definir la GEPA: asma, eosinofilia periférica mayor del 10%, anormalidad de senos paranasales, infiltrados pulmonares migratorios o transitorios, mononeuropatía o polineuropatía y una biopsia conteniendo un vaso sanguíneo en la que se identifique acumulación de eosinófilos en áreas extravasculares. Se necesita la presencia de 4 o más de estos criterios para establecer el diagnóstico, lo que proporciona una sensibilidad del 85% y una especificidad del 99,7%4. En el caso de nuestra paciente se cumplían 5 de estos criterios, además la biopsia de colon no solo revelaba infiltración eosinofílica extravascular, sino también datos de vasculitis. Los hallazgos de vasculitis pueden faltar bien porque las biopsias correspondan a zonas superficiales que no contienen vasos submucosos, bien porque los pacientes no han progresado de la fase eosinofílica a la vasculítica. Es importante también resaltar que las biopsias de zonas con afectación leve o incluso normales endoscópicamente pueden evidenciar infiltración eosinofílica que nos ayude a establecer el diagnóstico5.
En la GEPA la afectación del TGI se produce tanto en la fase eosinofílica como en la vasculítica, y puede estar afectado cualquier tramo del tracto digestivo. Los síntomas más comunes son el dolor abdominal (59%), diarrea (33%) y sangrado (18%). La afectación del intestino grueso está más descrita que la de intestino delgado (ID)6. En el pasado la afectación del ID se detectaba habitualmente durante una operación urgente para tratamiento de una perforación intestinal; en la actualidad el uso de técnicas como la cápsula endoscópica o la endoscopia de doble balón permiten el diagnóstico precoz de algunos casos de SCS que se presentan como ulceraciones en ID7,8. Existen formas de presentación más severas con isquemia, perforación u obstrucción intestinal que se asocian a mal pronóstico. En la GEPA la mortalidad secundaria a la afectación digestiva ocupa el cuarto lugar por detrás de la afectación cardíaca, neurológica y renal. El tratamiento de esta entidad se basa en el uso de corticoides (prednisona a dosis de 0,5 a 1,5mg/kg/día) durante 6 a 12 semanas con descenso gradual posterior. En las formas de presentación más severas puede ser preciso asociar inicialmente ciclofosfamida. Una vez inducida la remisión, como ocurría en nuestra paciente, el tratamiento de mantenimiento se basa en inmunosupresores menos tóxicos, como azatioprina o metotrexato, junto con dosis descendentes de corticoides durante 12 a 18 meses.
En conclusión, la GEPA es una vasculitis rara que puede afectar al TGI. La presentación con sintomatología digestiva es infrecuente y las manifestaciones clínicas pueden ir desde síntomas leves a manifestaciones graves. El diagnóstico de GEPA en el TGI requiere una elevada sospecha clínica, ya que puede faltar la documentación histológica de vasculitis y el único hallazgo orientativo puede ser la presencia de eosinofilia extravascular.

