





El síndrome de Cowden (SC) es una enfermedad genética rara caracterizada por el desarrollo de hamartomas que pueden afectar a múltiples órganos, con un riesgo incrementado de distintos tumores. Aunque la presencia de pólipos hamartomatosos es frecuente, es menos común la existencia de ganglioneuromas intestinales como manifestación inicial1,2.
Presentamos el caso de un paciente con poliposis ganglioneuromatosa en el que se llegó al diagnóstico final de SC a través del resto de manifestaciones extracolónicas.
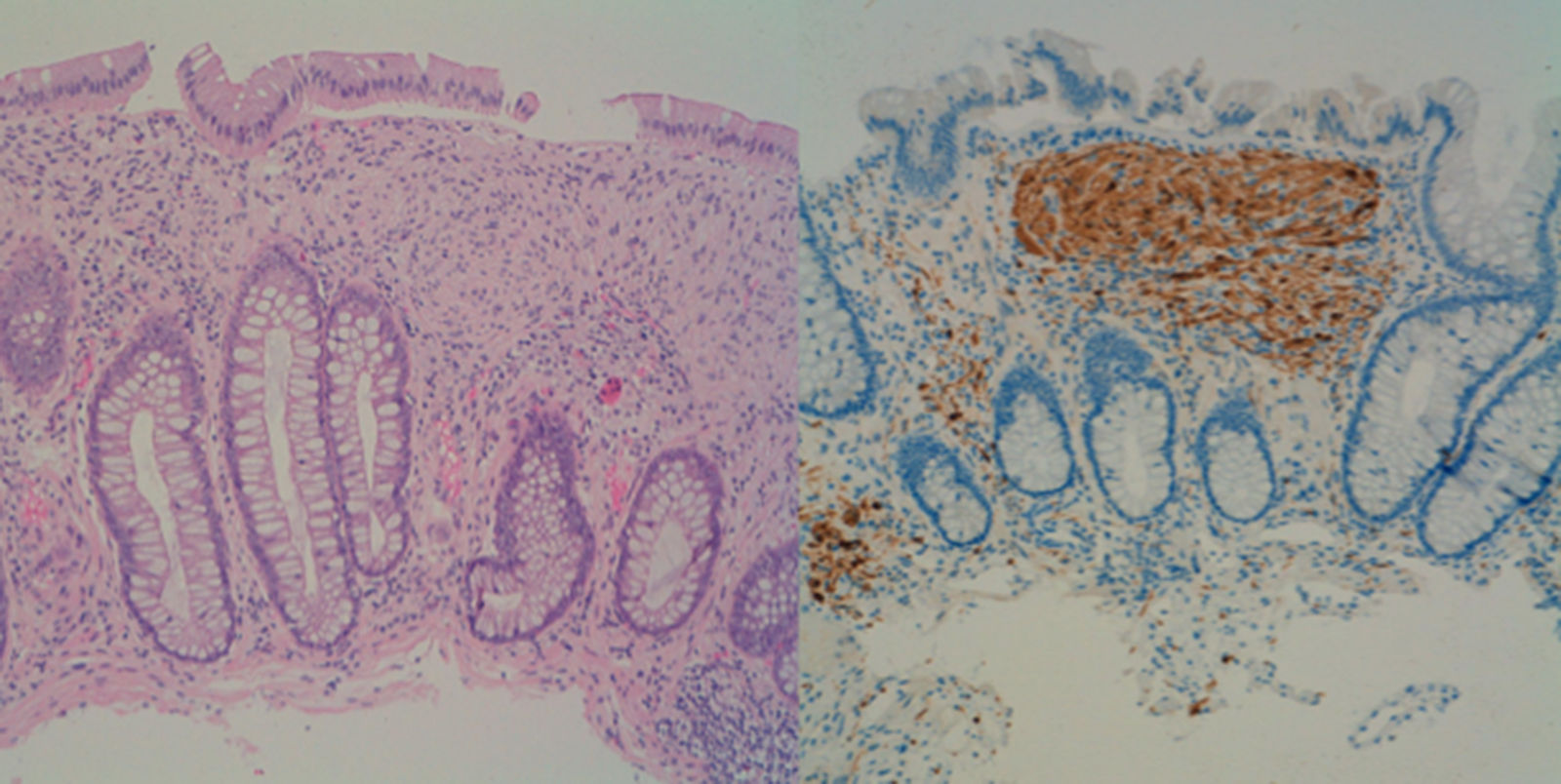
Se trata de un varón de 39 años que es remitido para la realización de colonoscopia por el antecedente de un familiar de primer grado fallecido de cáncer de colon (CCR). En la colonoscopia se objetivan más de 100 pólipos, distribuidos a lo largo de todo el colon, pero más frecuentes en el lado izquierdo. Tras la resección de varios de estos pólipos la histología muestra una proliferación fusocelular mesenquimal difusa con células ganglionares dispersas en lámina propia, todo ello compatible con pólipos ganglioneuromatosos (fig. 1). Ante estos hallazgos se solicita gastroscopia en la que se describe una extensa acantosis glucogénica en el esófago y más de 100 pólipos en la segunda porción duodenal, la mayoría milimétricos, que corresponden a adenomas tubulares con displasia moderada.

En la anamnesis dirigida destaca el antecedente de hemitiroidectomía por adenoma y la resección de un fibroma colágeno en el flanco derecho. A la exploración llama la atención una evidente macrocefalia (estudiado en la infancia y atribuido a raquitismo) y la presencia de máculas pigmentadas parduzcas en el pene.
Con todos estos hallazgos el paciente cumple criterios clínicos de SC3, por lo que se realiza un estudio de secuenciación completa del gen PTEN, obteniendo una mutación patogénica (p.Arg130Gln) en el exón 5 asociada a dicho síndrome. Se descartó mediante pruebas complementarias la asociación con neoplasia endocrina múltiple tipo ii.
La ganglioneuromatosis se debe a una hiperplasia de los plexos mientéricos y fibras nerviosas entéricas. Clínicamente se distinguen 3 entidades: 1) ganglioneuromatosis polipoidea, que consiste en pocos ganglioneuromas aislados; 2) ganglioneuromatosis generalizada, que se asocia a neoplasia endocrina múltiple tipo IIb y neurofibomatosis de von Recklinghausen; y 3) poliposis ganglioneuromatosa, sin enfermedad sistémica asociada, en la cual se encuadra el caso presentado2.
EL SC es una enfermedad hereditaria, con transmisión autosómica dominante, debida a mutación germinal en el gen supresor de tumores PTEN. Su prevalencia se estima en un caso por cada 200.000-250.000 habitantes4.
Inicialmente se propusieron unos criterios diagnósticos que incluían criterios patognomónicos, mayores y menores5. Dada su baja especificidad Pilarski et al. propusieron unos criterios alternativos que han sido adoptados por el National Comprehensive Cancer Network. Una de las principales diferencias es que se incluyen los hamartomas gastrointestinales, incluidos ganglioneuromas, como criterios mayores (tabla 1)3.
Criterios diagnósticos modificados del síndrome de Cowden
| Criterios mayores | Criterios menores |
|---|---|
| Cáncer de mama Cáncer de endometrio (epitelial) Hamartomas gastrointestinales (incluidos ganglioneuromas pero excluidos pólipos hiperplásicos≥3) Enfermedad de Lhermitte Duclos del adulto Macrocefalia (≥percentil 97) Pigmentación macular en el pene Múltiples lesiones mucocutáneas (cualquiera de las siguientes): Múltiples triquilemomas (≥3, al menos uno comprobado con biopsia) Queratosis acral (≥3placas queratósicas palmoplantares y/o pápulas hiperqueratósicas acrales) Neuromas mucocutáneos (≥3) Papilomas orales múltiples (particularmente en la lengua o en las encías), múltiples (≥3) o comprobado histológicamente o diagnosticado dermartológicamente | Espectro autista Cáncer de colon Acantosis glucogénica esofágica (≥3) Lipomas (≥3) Retraso mental (cociente intelectual ≤75) Carcinoma de células renales Lipomatosis testicular Cáncer de tiroides (papilar o folicular, variante de papilar) Lesiones estructurales de tiroides (adenoma, bocio multinodular…) Alteraciones vasculares (incluyen anomalías venosas intracraneales) |
El diagnóstico se establece en un individuo que cumpla cualquiera de estas 2 condiciones: 3 o más criterios mayores, pero alguno debe incluir macrocefalia, enfermedad de Lhermitte Duclos o hamartomas gastrointestinales o 2 criterios mayores y 3 menores.
En el caso de familias donde un individuo cumpla los criterios diagnósticos o tenga mutación en el gen PTEN, el diagnóstico se establece cuando se cumplan cualquiera de estas condiciones: a) 2 criterios mayores cualquiera con o sin criterios menores; o b) un criterio mayor y 2 menores; o c) 3 criterios menores.
Fuente: Pilarski et al.3.
Las lesiones mucocutáneas (98%), macrocefalia (93%), pólipos gastrointestinales (93%), lesiones benignas de mama (74%) y de tiroides (71%) son las manifestaciones más frecuentes6. Entre las expresiones digestivas más habituales destaca la acantosis glucogénica esofágica. A pesar de su inespecificidad se ha propuesto que la presencia de acantosis esofágica y poliposis colónica debe ser considerada como criterio patognomónico de SC. Se han descrito pólipos gastro-duodenales en un 67-100%, la mayoría hamartomatosos e hiperplásicos, pero también adenomas. Con respecto a la afectación colónica hasta un 92% tienen pólipos que varían desde pocos a innumerables. Es llamativa la coexistencia de múltiples tipos histológicos que incluyen hamartomas, pólipos hiperplásicos, ganglioneuromas o adenomas7.
Pero lo más importante es el alto riesgo de tumores malignos. Varios estudios coinciden en señalar un incremento de riesgo de cáncer de mama (25-50%), endometrial (13-28%), tiroideo (3-17%) o renal (2-5%)8,9. A diferencia de los que se creía previamente, también existe un mayor riesgo de CCR. Heald et al. describen que de 67 pacientes con SC sometidos a colonoscopia, un 13% presentaron CCR con una edad media de 44,4 años10. En un trabajo posterior el riesgo estimado fue 10,3 (IC 95%: 5,6-17,4, p<0,001) veces superior al de la población general8. Este índice disminuye a 5,7 (IC 95%: 1,14-16,64, p=0,036) en otro trabajo con 154 pacientes9. En un estudio multicéntrico que incluye 156 pacientes, el riesgo acumulativo de CCR fue del 18% a los 60 años11.
La importancia del diagnóstico precoz radica en la prevención de tumores asociados. Así como hay consenso en la vigilancia de las manifestaciones extraintestinales, el seguimiento endoscópico es más controvertido. La guía americana para el manejo de los síndromes hereditarios recomienda comenzar con colonoscopia a los 15 años, con una periodicidad bianual12. Sin embargo, otros autores proponen una vigilancia menos agresiva, comenzando a los 35-40 años con una periodicidad anual-bianual si existe alta tasa de pólipos o cada 3-5 años si existiera baja tasa de pólipos8,13. Así mismo, recomiendan la realización de gastroscopia en el momento del diagnóstico y con intervalos de 5 años o antes en función de los hallazgos10.
En conclusión, dado que en las poliposis colónicas pueden coexistir múltiples tipos histológicos, es importante valorar las manifestaciones extraintestinales que nos ayuden a determinar el diagnóstico final, ya que la vigilancia y manejo será diferente en cada caso.

