






La enfermedad inflamatoria intestinal (EII) es un trastorno de etiología desconocida consistente en una inflamación crónica del tubo digestivo. Los fármacos dirigidos contra el factor de necrosis tumoral han representado un hito en el tratamiento de los pacientes con EII en los últimos años contando además con un buen perfil de seguridad. No obstante, estos tratamientos no son eficaces en todos los pacientes y, en aquellos que responden inicialmente, se ha descrito una pérdida de respuesta a lo largo del tiempo. Por estos motivos, es necesario el desarrollo de nuevos tratamientos para la EII, dirigidos hacia diferentes dianas terapéuticas
En los últimos tiempos se han incorporado nuevas moléculas al arsenal terapéutico de los pacientes con EII. El golimumab es un anticuerpo monoclonal que se dirige contra el factor de necrosis tumoral y que ha demostrado ser eficaz en el tratamiento de la colitis ulcerosa. Asimismo, ha sido aprobado en Europa el uso de CT-P13 (infliximab biosimilar) para las mismas indicaciones que el infliximab original. Más recientemente, vedolizumab, un anticuerpo monoclonal dirigido frente a las integrinas α4β7 ha sido aprobado para el tratamiento de la enfermedad de Crohn y la colitis ulcerosa. En la actualidad se están desarrollando un gran número de moléculas, algunas de las cuales vendrán, en un futuro, a ampliar las opciones terapéuticas en los pacientes con EII.
Finalmente, en los próximos años los estudios deberán ir dirigidos a identificar factores predictores de respuesta a los distintos fármacos biológicos para la EII con el fin de seleccionar, de forma más personalizada, la mejor alternativa terapéutica para cada paciente.
Inflammatory bowel disease (IBD) is a disorder of unknown aetiology that provokes chronic inflammation of the gastrointestinal tract. Anti-tumor necrosis factor drugs have represented a major advance in the treatment of IBD patients in the last few years and also have a good safety profile. Nevertheless, these treatments are not effective in all patients and, in initial responders, there can be a loss of response in the long-term. Consequently, new treatments are needed for IBD, aimed at distinct therapeutic targets.
In the last few years, new molecules have been incorporated into the therapeutic armamentarium of IBD patients. Golimumab is an anti-tumor necrosis factor monoclonal antibody with demonstrated effectiveness in the treatment of ulcerative colitis. The use of CT-P13 (biosimilar infliximab) has been approved in Europe for the same indications as the original infliximab. More recently, vedolizumab, an anti-α4β7 integrin monoclonal antibody, has been approved for the treatment of Crohn's disease and ulcerative colitis. A large number of molecules are currently under development, some of which will, in the future, broaden the therapeutic options available in the treatment of IBD patients.
Finally, in the next few years, studies should aim to identify factors predictive of response to the distinct biological agents for IBD in order to allow personalized selection of the best therapeutic alternative for each patient.
La enfermedad inflamatoria intestinal (EII) es un trastorno crónico de etiología desconocida que implica una respuesta patológica tanto del sistema inmune innato como del adquirido que resulta en una inflamación crónica del tubo digestivo. La EII es el resultado de la interacción de distintos factores entre los que se encuentra la susceptibilidad genética, factores ambientales, agentes infecciosos, la flora entérica comensal y las alteraciones inmunológicas1,2. El amplio abanico de factores implicados en el desarrollo de la enfermedad así como la complejidad del sistema inmune ofrecen múltiples dianas terapéuticas, que se reflejan en la gran diversidad de moléculas que se han evaluado como potenciales tratamientos de la EII.
La era de la terapia biológica en el tratamiento de la EII se inició en 1998 cuando la Food and Drug Administration aprobó el infliximab para el tratamiento de los pacientes con enfermedad de Crohn3–5. Desde entonces, se han desarrollado y aprobado nuevos fármacos biológicos, hasta ahora todos dirigidos contra el factor de necrosis tumoral alfa (TNF). Los fármacos anti-TNF han representado un hito en el tratamiento de los pacientes con EII en los últimos años —disminuyendo la necesidad de cirugía y los ingresos hospitalarios y, lo más importante, mejorando la calidad de vida—, contando además con un buen perfil de seguridad. No obstante, estos tratamientos no son eficaces en todos los pacientes y, en aquellos que responden inicialmente, se ha descrito una pérdida de respuesta a lo largo del tiempo6–8. Además, aun siendo en general seguros, se asocian con la aparición de efectos adversos y tienen un elevado coste.
Por estos motivos, es necesario el desarrollo de nuevos tratamientos para la EII, dirigidos hacia diferentes dianas terapéuticas, buscando fármacos con un mecanismo de acción más específico para obtener un efecto más local en el órgano inflamado.
Recientemente, se han incorporado 3 nuevas moléculas al arsenal terapéutico de la EII: el golimumab y el CT-P13 (infliximab biosimilar), que son fármacos con mecanismo de acción anti-TNF, y el vedolizumab que inhibe la migración de los linfocitos a los tejidos del tubo digestivo a través del bloqueo de las integrinas α4β7.
El objetivo del presente capítulo es revisar, de una forma práctica y crítica, las moléculas biológicas recientemente aprobadas para el tratamiento de la EII, sus características, dosis, indicaciones y perfil de seguridad. Finalmente, presentaremos brevemente algunos de los grupos de moléculas en desarrollo para el tratamiento de la EII.
GolimumabEl golimumab (Simponi®, Janssen Biotech, Inc, Horsham, PA, EE. UU.) es un anticuerpo monoclonal contra el TNF que representa una nueva opción de tratamiento para los pacientes con colitis ulcerosa refractarios al tratamiento convencional. El golimumab fue aprobado en EE. UU. por la Food and Drug Administration en mayo de 2013 y más recientemente, en octubre de 2013, la European Medicines Agency (EMA) aprobó el golimumab para el tratamiento de los pacientes con colitis ulcerosa que han sido refractarios o con intolerancia a los tratamientos convencionales.
Mecanismo de acciónEl golimumab es un anticuerpo monoclonal transgénico (inmunoglobulina G1) que se dirige específicamente contra un epítopo de la molécula de TNF9,10. El golimumab se une tanto al TNF soluble como al transmembrana con una afinidad incluso superior al adalimumab y al infliximab11. Además, el golimumab presenta una mayor estabilidad conformacional y una mayor capacidad para inhibir citotoxicidad celular inducida por TNF y la activación de las células endoteliales humanas, en comparación con el infliximab y el adalimumab11.
Por otro lado, al igual que el infliximab y el adalimumab, el golimumab realiza su acción mediante la inducción de la apoptosis de las células inflamatorias al unirse al TNF transmembrana. A pesar de que cabría esperar un efecto similar al tratarse de una IgG completa, su capacidad para la inducción de la apoptosis a través del TNF transmembrana parece menor que en el caso del infliximab y el adalimumab. Por este motivo, el golimumab podría ser menos eficaz que el adalimumab y el infliximab en enfermedades granulomatosas, como la enfermedad de Crohn, en las que es muy relevante el efecto por la interacción del anticuerpo con el TNF transmembrana11. Finalmente, debido a que la concentración sérica del golimumab necesaria para neutralizar el TNF soluble es menor que en el caso de otros anti-TNF, se considera que es posible administrar las dosis de golimumab cada 4 semanas.
Eficacia del golimumab en la colitis ulcerosaEl estudio PURSUIT-SC induction evaluó la eficacia del golimumab en la inducción de la remisión en pacientes con colitis ulcerosa moderada-grave refractarios o con intolerancia al tratamiento con tiopurinas, esteroides o aminosalicilatos12. El estudio incluyó un subestudio en fase 2 de búsqueda de dosis y un subestudio en fase 3 de confirmación. Para ello se incluyeron pacientes con colitis ulcerosa con una puntuación entre 6 y 12 en el índice de Mayo, con al menos 2 puntos en el subíndice endoscópico. Los pacientes fueron aleatorizados a recibir placebo, golimumab 100mg/50mg (solo antes de la selección de la dosis), golimumab 200mg/100mg o golimumab 400mg/200mg por vía subcutánea en las semanas 0 y 2.
El objetivo primario fue evaluar la respuesta en la semana 6 después de la selección de dosis. Los objetivos secundarios fueron la remisión clínica, la cicatrización mucosa y la calidad de vida en la semana 6. Aunque para el análisis de seguridad se incluyeron todos los pacientes, para evaluar los objetivos del estudio no se tuvieron en cuenta los pacientes aleatorizados a recibir golimumab 100mg/50mg, ya que esta dosis se descartó en el subestudio en fase 2. Finalmente se analizaron los datos de 774 pacientes. Todos los objetivos del estudio fueron alcanzados por los pacientes tratados con golimumab en comparación con el grupo placebo (tabla 1).
Resultados de la eficacia del golimumab en la inducción de la respuesta en los pacientes con colitis ulcerosa moderada-grave en la semana 6 (estudio PURSUIT-SC induction)
| Placebo semanas 0 y 2 (N=256) (%) | Golimumab 200/100mg semanas 0 y 2 (N=257) (%) | Golimumab 400/200mg semanas 0 y 2 (N=258) (%) | |
|---|---|---|---|
| Respuesta | 29,7 | 51,8* | 55* |
| Remisión | 6,3 | 18,7* | 17,8* |
| Cicatrización mucosa | 28,5 | 43,2* | 45,3* |
N: número de pacientes.
El estudio PURSUIT-maintenance evaluó la eficacia del golimumab por vía subcutánea en el mantenimiento de la remisión en pacientes con colitis ulcerosa moderada-grave13. Se incluyeron pacientes que habían presentado respuesta en los estudios de inducción con golimumab. Los pacientes que habían respondido al tratamiento de inducción con golimumab fueron aleatorizados a recibir tratamiento con placebo, golimumab 50mg o golimumab 100mg cada 4 semanas hasta la semana 54. Los pacientes que habían recibido placebo y respondieron recibieron placebo de mantenimiento. Finalmente, los pacientes que no habían alcanzado respuesta con el golimumab o con placebo recibieron golimumab 100mg cada 4 semanas. El objetivo del estudio fue la respuesta a lo largo de las 54 semanas. Los objetivos secundarios se evaluaron en las semanas 30 y 54 y fueron la remisión clínica y la cicatrización mucosa.
De los pacientes que inicialmente respondieron al golimumab (464), el porcentaje de pacientes en remisión en la semana 54 fue significativamente más alto en los grupos que recibieron golimumab (50mg y 100mg) frente al grupo que recibió placebo (tabla 2).
Resultados de la eficacia del golimumab en el mantenimiento de la respuesta (semana 52) en los pacientes con colitis ulcerosa moderada-grave (estudio PURSUIT-maintenance)
| Placebo/4 semanas (N=156) (%) | Golimumab 50mg/4 semanas (N=153) (%) | Golimumab 100mg/4 semanas (N=154) (%) | |
|---|---|---|---|
| Respuesta | 31,4 | 47* | 50,6* |
| Remisión | 15,4 | 23,5** | 28,6* |
| Cicatrización mucosa | 26,9 | 41,8*** | 43,5* |
El golimumab se presenta en forma de jeringas precargadas que contienen 0,5mL de solución con 50mg de golimumab.
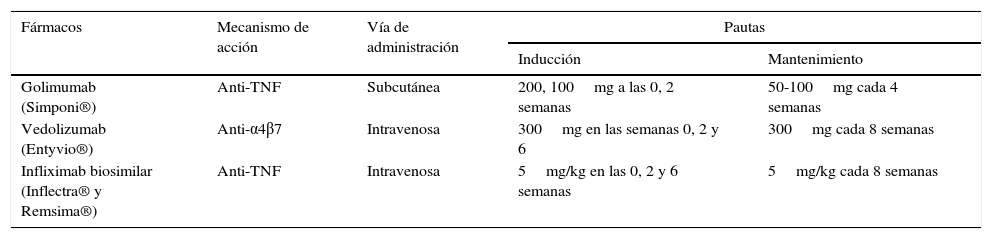
Dosis recomendada de golimumab para el tratamiento de la colitis ulcerosaSe recomienda la administración de 200mg de golimumab por vía subcutánea en la semana 0 y 100mg en la semana 2 seguidos de 50mg cada 4 semanas de mantenimiento. Para pacientes con peso igual o superior a 80kg se recomienda la administración de 100mg cada 8 semanas de mantenimiento (tabla 3).
Nuevos fármacos disponibles para la enfermedad inflamatoria intestinal, mecanismos de acción, vía de administración y dosis recomendadas
| Fármacos | Mecanismo de acción | Vía de administración | Pautas | |
|---|---|---|---|---|
| Inducción | Mantenimiento | |||
| Golimumab (Simponi®) | Anti-TNF | Subcutánea | 200, 100mg a las 0, 2 semanas | 50-100mg cada 4 semanas |
| Vedolizumab (Entyvio®) | Anti-α4β7 | Intravenosa | 300mg en las semanas 0, 2 y 6 | 300mg cada 8 semanas |
| Infliximab biosimilar (Inflectra® y Remsima®) | Anti-TNF | Intravenosa | 5mg/kg en las 0, 2 y 6 semanas | 5mg/kg cada 8 semanas |
El golimumab está indicado para el tratamiento de la colitis ulcerosa activa moderada-grave en pacientes adultos que han tenido una respuesta inadecuada al tratamiento convencional, incluidos corticosteroides e inmunosupresores, o que presenten intolerancia o contraindicaciones a dichas terapias.
SeguridadEn el estudio PURSUIT-SC induction, el porcentaje de efectos adversos fue similar en los pacientes expuestos al golimumab que en los que recibieron placebo12. Los acontecimientos adversos más frecuentes entre los pacientes tratados con golimumab fueron la cefalea y la nasofaringitis. Los efectos adversos graves fueron similares en los expuestos al golimumab que en los que recibieron placebo (3 vs. 6%). El porcentaje de infecciones graves también fue similar en los tratados con golimumab y en los que recibieron placebo (0,5 vs. 1,8%). Un paciente del grupo del golimumab 400mg/200mg falleció por complicaciones posquirúrgicas y otro paciente de ese mismo grupo presentó una enfermedad desmielinizante. El porcentaje de pacientes que precisó interrumpir el tratamiento debido a la aparición de efectos adversos fue bajo (el 0,5% en los tratados con golimumab y el 0,9% en los que recibieron placebo). La proporción de pacientes con reacciones en el punto de punción fue bajo (el 3,4% en los grupos de golimumab y el 1,5% en el grupo placebo), sin que hubiera diferencias en función de la dosis de golimumab.
En el estudio PURSUIT-maintenance el golimumab demostró un buen perfil de seguridad13. La tasa de efectos adversos, efectos adversos graves, infecciones e infecciones graves fueron similares entre los distintos grupos de tratamiento. Del mismo modo, la frecuencia de abandono del tratamiento por efectos secundarios fue similar en los 3 grupos de estudio. El porcentaje de pacientes con reacciones en el punto de punción fue similar en los grupos tratados con golimumab y en el grupo placebo.
VedolizumabEl vedolizumab (Entyvio®, Millennium Pharmaceuticals, Cambridge, MA, EE. UU.) es un anticuerpo monoclonal humanizado que se une específicamente al heterodímero α4β7, bloqueando de forma selectiva la migración de los linfocitos al intestino, si afectar su migración a otros órganos como el sistema nervioso central (SNC). Durante el desarrollo de este fármaco la molécula sufrió modificaciones, recibiendo distintos nombres (LDP02 y MLN0002) hasta la versión actual conocida como MLN0002 o vedolizumab14.
Mecanismo de acciónLa EII supone una inflamación crónica en el tubo digestivo para lo cual es necesaria la migración de las células mediadoras de la inflamación a los tejidos afectos. La migración de los leucocitos a los tejidos intestinales inflamados es un proceso que está regulado por complejos mecanismos moleculares. Las integrinas α4β7 son glucoproteínas que se activan en la superficie de los linfocitos B y T circulantes e interaccionan con las moléculas de adhesión celular de adresina mucosa 1 (MAdCAM-1) del endotelio de los vasos. MAdCAM-1 se expresa selectivamente en el endotelio de los vasos intestinales, uniendo los linfocitos que circulan a gran velocidad en el torrente sanguíneo15. Así, estos linfocitos migran desde la superficie endotelial a la lámina propia y los tejidos. La permanencia en número excesivo de estos linfocitos en los tejidos intestinales forma parte del proceso fisiopatogénico de la EII16. Distintos estudios han observado un aumento significativo de la expresión de α4β7 y MadCAM-1 en el colon de los pacientes con colitis ulcerosa en comparación con los pacientes con síndrome de intestino irritable17.
El natalizumab, la molécula predecesora del vedolizumab, demostró el potencial de este mecanismo de acción en estudios fase 2. El natalizumab es un anticuerpo monoclonal que ha demostrado ser eficaz tanto en la esclerosis múltiple como en la enfermedad de Crohn y que se une a la subunidad α4 de las integrinas bloqueando, por tanto, la interacción de los heterodímeros α4β1 y α4β7 con sus receptores, incluyendo fibronectina, VCAM-1 y MadCAM-118–20. A pesar de su eficacia, el natalizumab fue retirado del mercado en 2005 por la aparición de leucoencefalopatía multifocal progresiva (LMP), que es una infección grave del SNC asociada al tratamiento con este fármaco. En 2006 fue introducido de nuevo para ser dispensado bajo programas de vigilancia y actualmente está aprobado en EE. UU., aunque no en Europa, para la enfermedad de Crohn. Desde su reintroducción, 52.000 pacientes han recibido natalizumab en monoterapia, la mayoría para el tratamiento de la esclerosis múltiple, y se han comunicado 10 nuevos casos de LMP21. Se cree que la LMP se debe a la reactivación del virus de John Cunnigham en situaciones de inmunodepresión, lo que favorece la diseminación del virus por el torrente circulatorio. Debido a la unión del natalizumab a las integrinas α4β1, se encuentra bloqueada la migración de los linfocitos al SNC, lo que impide el aclaramiento del virus del SNC por el sistema inmune. La diferencia entre el vedolizumab y el natalizumab es que el segundo bloquea la migración de los linfocitos a múltiples órganos (como el tubo digestivo y el SNC), mientras que el vedolizumab actúa específicamente en el tubo digestivo. Estudios en primates han mostrado que el vedolizumab es específico del tracto digestivo y que no se asocia con alteraciones en el líquido cefalorraquídeo22. Además, en un estudio llevado a cabo en un pequeño grupo de voluntarios sanos que se sometieron a una punción lumbar antes y después de una dosis de 450mg de vedolizumab no se observaron diferencias en CD4+ y CD8+ o el ratio de ellos23.
En 1996 se empleó por primera vez un anticuerpo monoclonal contra α4β7, demostrando su capacidad para controlar la colitis en modelos animales24. Los monos tamarindos, cuando están en cautividad, desarrollan de forma espontánea una colitis crónica que se asemeja a la colitis ulcerosa. Los monos incluidos en el estudio fueron diagnosticados de colitis por criterios endoscópicos e histológicos y aleatorizados a recibir el fármaco anti-α4β7 o un anticuerpo monoclonal no terapéutico por vía intramuscular. Los primates que recibieron el anticuerpo anti-α4β7 presentaron una rápida mejoría clínica, endoscópica e histológica que no se observó en los animales no tratados24.
Eficacia del vedolizumabEl primer estudio con un anti-α4β7 en humanos se publicó en el año 2000 en forma de resumen. Se trató de un estudio doble ciego controlado con placebo en el que se incluyeron 29 pacientes con colitis ulcerosa moderada-grave. Los pacientes fueron aleatorizados a recibir una dosis del anticuerpo humanizado (LDP02) en distintas dosis: 0,15mg/kg subcutáneo, 0,15mg/kg intravenoso, 0,5mg/kg intravenoso, 3mg/kg intravenoso o placebo. Una sola dosis de 0,5mg/kg fue suficiente para saturar completamente los receptores de los anticuerpos25.
En un estudio en fase 1 realizado en pacientes con colitis ulcerosa, Feagan et al. demostraron que, tras una única infusión de vedolizumab (entonces denominado MLN0002), el bloqueo de las integrinas α4β7 fue superior al placebo en la inducción clínica y endoscópica25. El primer estudio fase 2 se realizó en pacientes con colitis ulcerosa naïve al tratamiento biológico. Se incluyeron 181 pacientes con colitis ulcerosa que fueron aleatorizados a recibir MLN0002 a dosis de 2mg/kg, 0,5mg/kg o placebo en las semanas 0 y 426. El objetivo primario fue la remisión en la semana 6. En ambos grupos (2 y 0,5mg/kg), los porcentajes de remisión clínica fueron superiores en los pacientes tratados con MLN0002 que en los expuestos a placebo (33, 32 y 14% respectivamente, p=0,03). Del mismo modo, el porcentaje de pacientes que alcanzó la remisión endoscópica fue significativamente más alto en los tratados con MLN0002 (2 y 0,5mg/kg) que con placebo (28, 12 y 8%, respectivamente)26.
En un segundo estudio en fase 2 se incluyeron 185 pacientes con enfermedad de Crohn naïve para tratamiento anti-TNF que se aleatorizaron a recibir MLN0002 a dosis de 2mg/kg, 0,5mg/kg o placebo en las semanas 0 y 427. En la semana 6, el 37 y el 30% de los pacientes tratados con 2 y 0,5mg/kg de MLN0002, respectivamente, alcanzaron la remisión, en comparación con el 21% de los pacientes tratados con placebo. El porcentaje de efectos adversos fue similar entre los grupos27.
A pesar de la eficacia del anticuerpo anti-α4β7 demostrada en estos estudios, aproximadamente el 40% de los pacientes desarrolló anticuerpos contra el fármaco26. Además, una concentración superior a 1:125 de anticuerpos contra el fármaco se asoció con probabilidad de respuesta más baja debido a una menor saturación de las integrinas α4β726. Por esto, Millennium Pharmaceuticals desarrolló una nueva formulación del anticuerpo (el inicial era producido por la línea celular N0 de mieloma de ratón y el nuevo por un sistema celular basado en ovario de hámster chino) que fue evaluada en un estudio de búsqueda de dosis (2, 6 y 10mg/kg)28. En este estudio, la concentración sérica del vedolizumab (MLN0002) aumentó proporcionalmente con la dosis administrada y los receptores α4β7 de los linfocitos de sangre periférica se saturaron con cualquiera de las dosis evaluadas. Posteriormente, en un estudio abierto en fase 2 incluyendo a pacientes con enfermedad de Crohn y colitis ulcerosa se evaluó la nueva formulación con las mismas dosis (2, 6 y 10mg/kg) pero administradas con mayor frecuencia (en las semanas 0, 2 y 6 en lugar de las semanas 0 y 4, como en los primeros estudios en fase 2)29. En este estudio tan solo el 4% de los pacientes desarrolló anticuerpos contra el vedolizumab. Los autores concluyeron que la combinación del cambio de formulación, la dosis más alta y la administración más frecuente del fármaco contribuyeron a una menor proporción de formación de anticuerpos.
Más recientemente, el vedolizumab ha sido evaluado en estudios fase 3 (GEMINI I, II, III y LTS)30–33. El estudio GEMINI I evaluó la eficacia del vedolizumab en la inducción y el mantenimiento de la remisión en pacientes con colitis ulcerosa moderada-grave30. La respuesta clínica se definió como una disminución de al menos 3 puntos en el índice de Mayo y una disminución de al menos un 30% con respecto a la basal, con una diminución en la puntuación de «sangrado rectal» de al menos un punto con una puntuación en el subíndice de «sangrado rectal»≤1. La remisión clínica se definió como una puntuación de≤2 en el índice de Mayo con una puntuación≤1 en todos los subíndices. En la fase de inducción, 374 pacientes fueron aleatorizados a recibir vedolizumab 300mg en las semanas 0 y 2 o placebo (cohorte 1) y 520 pacientes recibieron vedolizumab 300mg en las semanas 0 y 2 en abierto (cohorte 2). En el estudio de mantenimiento, los pacientes que en la semana 6 habían respondido al vedolizumab fueron aleatorizados a continuar con vedolizumab 300mg/8 semanas, vedolizumab 300mg/4 semanas o placebo hasta la semana 52.
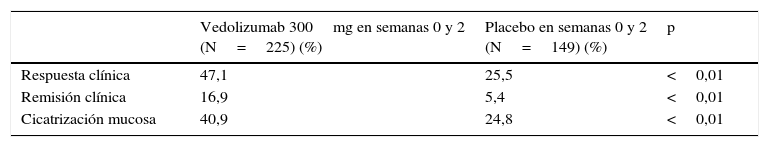
El porcentaje de pacientes que presentó respuesta en la semana 6 fue superior en los pacientes tratados con vedolizumab que en los que recibieron placebo (47 vs. 25,5%). Además, el porcentaje de pacientes que alcanzó la remisión y el porcentaje de pacientes con cicatrización mucosa fue más alto en los pacientes tratados con vedolizumab que en los que recibieron placebo (tabla 4).
Resultados de la eficacia del vedolizumab en la inducción de la remisión (semana 6) en pacientes con colitis ulcerosa (estudio GEMINI I)
| Vedolizumab 300mg en semanas 0 y 2 (N=225) (%) | Placebo en semanas 0 y 2 (N=149) (%) | p | |
|---|---|---|---|
| Respuesta clínica | 47,1 | 25,5 | <0,01 |
| Remisión clínica | 16,9 | 5,4 | <0,01 |
| Cicatrización mucosa | 40,9 | 24,8 | <0,01 |
N: número de pacientes.
Fuente: Feagan et al.30.
En la semana 52, el porcentaje de pacientes en remisión fue significativamente más alto en los que recibieron vedolizumab que en los aleatorizados a placebo. Además, el porcentaje de pacientes con respuesta clínica sostenida, cicatrización mucosa y remisión libre de esteroides fue significativamente más alto en los pacientes aleatorizados a vedolizumab que en los que recibieron placebo. En el análisis post-hoc, la dosis de vedolizumab 300mg/4 semanas no fue superior a la dosis de 300mg/8 semanas. El tratamiento concomitante con esteroides o con inmunosupresores no se asoció con una mayor eficacia del fármaco. Además, el fracaso previo al tratamiento con anti-TNF no se asoció con una menor eficacia del vedolizumab.
Se observó correlación entre los niveles de vedolizumab y la respuesta. La concentración media de vedolizumab con la dosis de 300mg/4 semanas fue mayor que en los pacientes con 300mg/8 semanas (38,3 vs. 11,2μg/mL). Sin embargo, en ambos casos la saturación de los α4β7 de los linfocitos fue superior al 95%.
El 3,7% de los pacientes desarrollaron anticuerpos contra vedolizumab. El tratamiento concomitante con inmunosupresores disminuyó la incidencia de formación de anticuerpos contra el vedolizumab.
El estudio GEMINI II evaluó la eficacia del vedolizumab en la inducción y el mantenimiento de la remisión en pacientes con enfermedad de Crohn refractarios a tratamientos convencionales o a anti-TNF31. Se incluyeron pacientes con enfermedad de Crohn moderada-grave definida con un CDAI de 220-450 junto con algún dato objetivo de inflamación: PCR elevada, o actividad endoscópica (≥3 úlceras grandes o ≥10 aftas) o calproptectina fecal≥250μg/g y evidencia de úlceras en la tomografía computarizada, resonancia magnética, tránsito intestinal o cápsula endoscópica). Los objetivos primarios de la fase de inducción fueron la remisión clínica definida como un CDAI≤150 en la semana 6 y la respuesta clínica definida como un descenso≥100 puntos con respecto al CDAI en la semana 0.
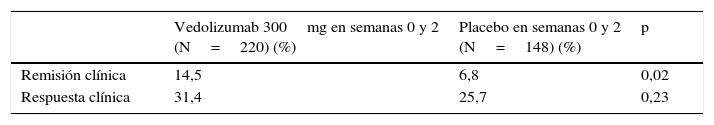
En la fase de inducción, 368 pacientes fueron aleatorizados a recibir vedolizumab 300mg o placebo en las semanas 0 y 2 (cohorte 1) y 747 pacientes recibieron vedolizumab 300mg de forma abierta en las semanas 0 y 2. En la semana 6, el porcentaje de pacientes que alcanzó la remisión fue significativamente más alto en los tratados con vedolizumab que en los que recibieron placebo. Sin embargo, el porcentaje de respuesta fue similar en ambos grupos (tabla 5).
Resultados de la eficacia del vedolizumab en la inducción de la remisión (semana 6) en pacientes con enfermedad de Crohn (estudio GEMINI II)
| Vedolizumab 300mg en semanas 0 y 2 (N=220) (%) | Placebo en semanas 0 y 2 (N=148) (%) | p | |
|---|---|---|---|
| Remisión clínica | 14,5 | 6,8 | 0,02 |
| Respuesta clínica | 31,4 | 25,7 | 0,23 |
Fuente: Sandborn et al.31.
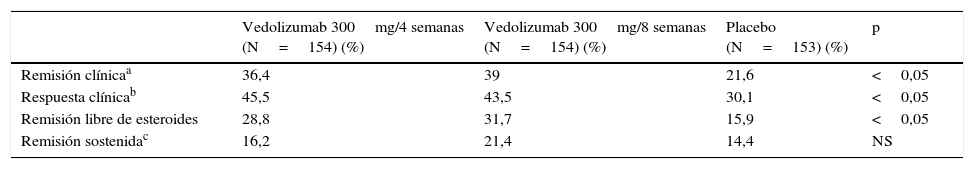
En la semana 6, los 461 pacientes que habían presentado respuesta con el tratamiento con vedolizumab fueron aleatorizados a recibir vedolizumab 300mg/4 semanas, vedolizumab 300mg/8 semanas o placebo hasta la semana 52. El objetivo primario de la fase de mantenimiento fue la remisión clínica en la semana 52 (CDAI≤150). En la semana 52, el porcentaje de remisión en los pacientes que recibieron vedolizumab 300mg tanto cada 4 como cada 8 semanas fue significativamente más alto en el grupo placebo (tabla 6).
Resultados de la eficacia del vedolizumab en el mantenimiento de la remisión (semana 52) en pacientes con enfermedad de Crohn (estudio GEMINI II)
| Vedolizumab 300mg/4 semanas (N=154) (%) | Vedolizumab 300mg/8 semanas (N=154) (%) | Placebo (N=153) (%) | p | |
|---|---|---|---|---|
| Remisión clínicaa | 36,4 | 39 | 21,6 | <0,05 |
| Respuesta clínicab | 45,5 | 43,5 | 30,1 | <0,05 |
| Remisión libre de esteroides | 28,8 | 31,7 | 15,9 | <0,05 |
| Remisión sostenidac | 16,2 | 21,4 | 14,4 | NS |
N:número de pacientes; NS: no estadísticamente significativo.
La respuesta clínica se definió como un descenso del CDAI≥100 puntos en la semana 52 con respecto a la basal.
La remisión clínica sostenida se definió como un CDAI≤150 en el 80% de las visitas del estudio, incluida la última (semana 52).
Fuente: Sandborn et al.31.
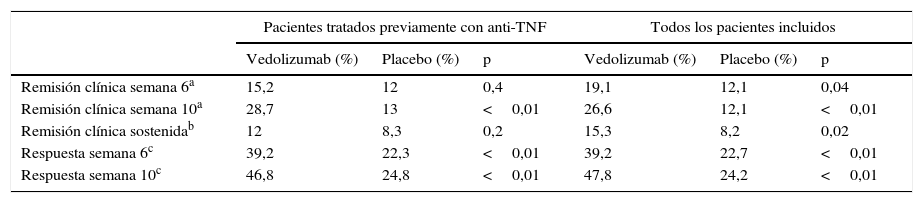
El estudio GEMINI III es un estudio aleatorizado, doble ciego, controlado con placebo que evaluó la seguridad y la eficacia de vedolizumab en la inducción de la remisión en 416 pacientes con enfermedad de Crohn refractarios a otros tratamientos34. El objetivo primario, que fue demostrar la eficacia del vedolizumab en la inducción de la remisión en la semana 6 en el grupo de pacientes que había fracasado previamente en el tratamiento con anti-TNF, no se alcanzó en este estudio. Sin embargo, la respuesta clínica en la semana 6 (descenso en CDAI≥100 puntos con respecto a la puntuación basal) y la remisión en la semana 10 fueron mayores en los pacientes tratados con vedolizumab (tabla 7).
Eficacia del vedolizumab en la inducción de la remisión en los pacientes con enfermedad de Crohn (GEMINI III)
| Pacientes tratados previamente con anti-TNF | Todos los pacientes incluidos | |||||
|---|---|---|---|---|---|---|
| Vedolizumab (%) | Placebo (%) | p | Vedolizumab (%) | Placebo (%) | p | |
| Remisión clínica semana 6a | 15,2 | 12 | 0,4 | 19,1 | 12,1 | 0,04 |
| Remisión clínica semana 10a | 28,7 | 13 | <0,01 | 26,6 | 12,1 | <0,01 |
| Remisión clínica sostenidab | 12 | 8,3 | 0,2 | 15,3 | 8,2 | 0,02 |
| Respuesta semana 6c | 39,2 | 22,3 | <0,01 | 39,2 | 22,7 | <0,01 |
| Respuesta semana 10c | 46,8 | 24,8 | <0,01 | 47,8 | 24,2 | <0,01 |
La respuesta clínica se definió como un descenso del CDAI≥100 puntos con respecto a la basal.
Fuente: Sands et al.34.
Finalmente, actualmente se está llevando a cabo un estudio de extensión abierto para evaluar la seguridad a largo plazo del vedolizumab GEMINI LTS35. Los resultados preliminares apoyan la seguridad del vedolizumab a largo plazo en el tratamiento de la enfermedad de Crohn y la colitis ulcerosa. Los resultados del estudio completo (a 7 años de seguimiento) estarán disponibles en 2016.
Modo de presentaciónLos envases contienen 300mg de vedolizumab polvo concentrado para solucio¿n para perfusio¿n.
PosologíaLa pauta posolo¿gica recomendada de Entyvio® es de 300mg administrados mediante perfusio¿n intravenosa en las semanas 0, 2 y 6, y cada 8 semanas a partir de entonces (tabla 3). Debe valorarse interrupir el tratamiento en los pacientes que no hayan presentado respuesta tras 14 semanas de tratamiento. En los pacientes que pierdan la respuesta inicial a lo largo del tiempo podría aumentarse la frecuencia de administración a 300mg/4 semanas.
IndicacionesEl vedolizumab está indicado en los pacientes con enfermedad de Crohn o colitis ulcerosa activa, de moderada a grave, en pacientes adultos que hayan tenido una respuesta inadecuada, presenten pérdida de respuesta o sean intolerantes al tratamiento convencional o a un anti-TNF.
SeguridadEn general, el porcentaje de efectos secundarios en los pacientes tratados con vedolizumab ha sido similar a los descritos en el grupo placebo. No se han descrito casos de LMP. No obstante, futuros estudios darán información sobre el perfil de seguridad del vedolizumab a largo plazo.
CT-P13El precio de los fármacos biológicos junto con su uso cada vez más extendido plantea un dilema por los costes asociados a estos tratamientos. Este hecho ha llevado a algunas agencias nacionales a restringir su uso tras un tiempo después de haber alcanzado la remisión36,37. El desarrollo de genéricos de fármacos constituidos por pequeñas moléculas ha demostrado permitir reducciones de precio del 80% comparado con las marcas originales38. Del mismo modo que ocurre con los demás fármacos, cuando las patentes de los biológicos están próximas a caducar comienzan a desarrollarse «copias» de las moléculas originales que en este caso se conocen como biosimilares. El desarrollo de fármacos biosimilares se plantea como una oportunidad para disponer de fármacos biológicos similares a los originales pero con un menor precio.
Un biosimilar es una copia de un fármaco biológico aprobado cuya patente ha expirado39. Sin embargo, mientras que un fármaco genérico es una copia exacta de una molécula de pequeño tamaño y relativamente poca complejidad, el biosimilar no es idéntico a la molécula original, sino que puede ser distinta por diferencias en el proceso de fabricación, el tipo de cultivo celular empleado para su síntesis, el proceso de purificación, la formulación o las condiciones de almacenamiento. Estos cambios podrían ser responsables de diferencias en la calidad, eficacia y seguridad del tratamiento, especialmente por cambios en la inmunogenicidad40. Este aspecto no es exclusivo de los biosimilares, sino que distintos lotes de los biológicos originales pueden presentar diferencias por cambios en el proceso de fabricación a lo largo del tiempo. En ocasiones, como consecuencia de estos cambios en el proceso de producción, los fabricantes de los biológicos originales tienen que demostrar su comparabilidad a las autoridades reguladoras41.
El nombre «biosimilar» fue propuesto por la EMA, que fue la primera autoridad reguladora en establecer cuál debía ser el proceso para la aprobación de estos fármacos. Tras el primer fármaco biosimilar aprobado por la EMA en 2005, 16 biosimilares han sido autorizados hasta ahora en Europa.
El proceso para la aprobación de un fármaco biosimilar es diferente al que se sigue para el desarrollo de un fármaco original41. La EMA establece que los fabricantes de la molécula biosimilar tienen que demostrar comparabilidad con la original en 3 aspectos: características físico-químicas de las moléculas, comparabilidad no clínica (evaluando los teóricos mecanismos de acción en distintos modelos) y comparabilidad clínica, que deben realizarse para la indicación en la que la población de pacientes es más sensible42. Aunque puede haber pequeñas diferencias, el proceso es bastante similar en Japón, Australia, Canadá y EE. UU.
Tras un complejo proceso de evaluación de los datos aportados por el fabricante de la molécula biosimilar, el producto será aceptado si se considera «altamente similar». Una vez demostrada esta alta similitud, los requerimientos de estudios clínicos son menos exigentes que para el producto de referencia, asumiendo que la alta similitud también predice que los resultados clínicos serán altamente similares42.
Comparabilidad del CT-P13 con el Remicade®En 2013 la EMA aprobó el primer biosimilar de Remicade® (infliximab) desarrollado por Celltrion como CT-P13 y comercializado como Remsima® por Celltrion e Inflectra® por Hospira42.
Los estudios clínicos con este fármaco se realizaron en artritis reumatoide y espondilitis anquilosante43,44. Además de para estas indicaciones, la EMA aprobó el uso del CT-P13 para todas las indicaciones del Remicade® (lo que se conoce con el nombre de extrapolación)42.
El CT-P13 tiene una secuencia de aminoácidos idéntica a la del Remicade®, ya que este es un requisito indispensable para ser considerado similar. La caracterización físico-química del CT-P13 demostró que el producto es altamente similar al Remicade®. Las distintas funciones del CT-P13 evaluadas, incluida la afinidad de unión con el TNF, su efecto de supresión de la producción de citoquinas en varias líneas celulares humanas y el resto de análisis llevados a cabo mostraron resultados que se encontraban dentro de los parámetros descritos para el Remicade®. La única diferencia observada entre el Remicade® y el CT-P13 fue en la proporción de formas afucosiladas, lo que se tradujo en una menor afinidad de unión de CT-P13 con el receptor FcγRIIIa y en una menor actividad de citotoxicidad celular mediada por anticuerpo en las células natural killers en experimentos in vitro. Sin embargo, cuando este experimento se realizó en condiciones más fisiológicas estas diferencias desaparecieron, mostrando ambos, el Remicade® y el CT-P13, una similar actividad de citotoxicidad42.
El desarrollo del primer infliximab biosimilar ha originado un gran debate sobre los requisitos necesarios para la aprobación de un producto biosimilar en todas las indicaciones del producto de referencia. Las guías europeas establecen que, si está convenientemente justificado, los biosimilares deben recibir la autorización para todas las indicaciones del producto de referencia, lo que se conoce como extrapolación de las indicaciones42.
El argumento que apoya la extrapolación es que el objetivo de los estudios para el desarrollo de los biosimilares no es demostrar eficacia en las distintas indicaciones per se, que ya ha sido demostrada por el fármaco original, sino que la similitud demostrada en los estudios clínicos, preclínicos y farmacocinéticos traducen una alta probabilidad de resultados clínicos comparables.
Varias sociedades científicas han adoptado una posición en contra de la extrapolación ya que consideran que son necesarios datos clínicos para conocer la eficacia del biosimilar en todas las indicaciones45,46. Otras, por el contrario, han apoyado la extrapolación y han optado por una estricta monitorización postautorización del biosimilar47. Los detractores de la extrapolación de las indicaciones a la EII argumentan que las diferencias en la afucosilación pueden afectar el efecto del fármaco en esta enfermedad. De hecho se han observado diferencias entre la artritis reumatoide y la EII en cuanto a la respuesta al tratamiento con otros anticuerpos monoclonales: algunos fármacos han demostrado ser eficaces en algunas enfermedades y no en otras, otros han demostrado ser activos en ambas enfermedades pero a diferentes dosis45,46.
Eficacia del CT-P13 en los estudios clínicosLos estudios para la aprobación del CT-P13 se han realizado en pacientes con artritis reumatoide y espondilitis anquilosante. El objetivo del estudio PLANETRA fue comparar la eficacia y seguridad del Remicade® con el CT-P13 en pacientes con artritis reumatoide43. El objetivo primario fue evaluar la respuesta en la semana 30 definida como una disminución en la American College of Rheumatology del 20%. El CT-P13 demostró una eficacia equivalente al Remicade® en la semana 30, con un perfil similar de farmacocinética e inmunogenicidad. El CT-P13 fue bien tolerado y con un perfil de seguridad comparable al Remicade®.
Por otro lado, el objetivo del estudio PLANETAS fue comparar la farmacocinética, seguridad y eficacia del Remicade® con el CT-P13 en pacientes con espondilitis anquilosante44. El objetivo primario fue evaluar el área bajo la curva de la concentración del fármaco así como la concentración máxima a las semanas 22 y 30 de tratamiento. Como objetivos secundarios se evaluó la respuesta y la seguridad de ambos tratamientos. Este estudio demostró que los perfiles farmacocinéticos del Remicade® y del CT-P13 fueron similares en pacientes con espondilitis anquilosante. El CT-P13 fue bien tolerado, con un perfil de seguridad y eficacia comparable al de Remicade® al menos hasta la semana 30.
Una de las cuestiones que plantean los detractores de la extrapolación de las indicaciones del CT-P13 es la conveniencia de las enfermedades elegidas para los estudios clínicos. Los pacientes con artritis reumatoide son el grupo más numeroso de pacientes que reciben tratamiento con los fármacos anti-TNF, pero desde el punto de vista de obtener la aprobación de las autoridades reguladoras no parece la población más sensible.
Para obtener la aprobación del fármaco, debe aportarse un plan de manejo de riesgos para cada anticuerpo biosimilar. Además de la farmacovigilancia habitual, el plan de manejo de la EMA del CT-P13 incluye varios estudios de extensión postautorización que en total incluirán más de 6.000 pacientes, entre los cuales se encuentra un ensayo clínico aleatorizado comparando la eficacia del CT-P13 con el Remicade® en pacientes con enfermedad de Crohn. A pesar de que la EMA apoya con argumentos sólidos la extrapolación de las indicaciones del Remicade®, el disponer de los resultados de un ensayo clínico comparativo de la eficacia del CT-P13 y del Remicade® en enfermedad de Crohn apoyará y facilitará la aceptación del uso del biosimilar en EII.
Moléculas en desarrollo para el tratamiento de la enfermedad inflamatoria intestinalLa investigación sobre los mecanismos fisiopatogénicos implicados en la EII ha llevado parejo el descubrimiento de potenciales dianas terapéuticas para el desarrollo de nuevos fármacos para el tratamiento de la EII. La mayoría de las moléculas en investigación tratan de bloquear la activación de los linfocitos T, la adhesión leucocitaria o inhibir la producción o el efecto de citoquinas proinflamatorias.
Bloqueo de citoquinas proinflamatoriasFactor de necrosis tumoralEl TNF-kinoide es una vacuna cuyo fin es inducir la producción de anticuerpos policlonales neutralizantes contra el TNF por el propio paciente. En un estudio abierto fase 1-2 en pacientes con enfermedad de Crohn se evaluaron 3 dosis de TNF-kinoide en 22 pacientes48. No se observaron acontecimientos adversos graves y todos los pacientes completaron el estudio. Los resultados preliminares del ensayo clínico en fase 2 no han sido tan alentadores49.
HMPL-004Andrographis paniculata es una planta considerada medicinal; se ha descrito que el extracto de esta planta podría poseer propiedades antiinflamatorias mediante la inhibición de la síntesis de óxido nítrico y de la vía NF-κβ50. Además, ha demostrado tener un efecto inhibidor de la activación de las células del sistema inmune.
HMPL-004 es uno de los componentes del extracto de Andrographis paniculata que ha demostrado resultados prometedores en el tratamiento de la EII. Esta molécula está siendo actualmente evaluada en estudios fase 3 en enfermedad de Crohn y colitis ulcerosa.
Interleucinas-12 y -23Las interleucinas-12 y -23 son citoquinas proinflamatorias que comparten una subunidad común (p40). El ustekinumab es un anticuerpo monoclonal IgG1 que se une a la subunidad p40 de las interleucinas-12 y -23 y ha demostrado ser eficaz en el tratamiento de la artritis psoriásica y de la psoriasis51–53.
La eficacia del ustekinumab en la inducción de la remisión en pacientes con enfermedad de Crohn fue evaluada por primera vez en un ensayo clínico doble ciego controlado con placebo por Sandborn et al.54. El grupo de pacientes tratados con ustekinumab presentó un mayor porcentaje de respuesta clínica en las semanas 4 y 6 que los tratados con placebo; no obstante, no se observaron diferencias en la semana 8.
Posteriormente, la eficacia del ustekinumab en la inducción y el mantenimiento de la remisión en pacientes con enfermedad de Crohn fue evaluada por Sandborn et al. en un estudio fase 2b55. Se incluyeron pacientes con enfermedad de Crohn moderada-grave refractarios al tratamiento con anti-TNF. Los pacientes fueron aleatorizados a recibir ustekinumab 1, 3 o 6mg/kg por vía intravenosa o placebo en la semana 0. La respuesta clínica en la semana 6 fue significativamente mayor en los tratados con ustekinumab que en los que recibieron placebo. Sin embargo, no se observaron diferencias en la remisión. Cabe destacar que, aunque el objetivo primario se estableció en la semana 6, los porcentajes de pacientes tratados con ustekinumab que alcanzaron respuesta o remisión en la semana 8 fueron superiores a los de la semana 6, mostrando que el mecanismo de acción es más lento y que quizás la semana 6 es demasiado precoz para valorar la respuesta.
Los pacientes respondedores a ustekinumab en la semana 6 fueron aleatorizados de nuevo a recibir ustekinumab 90mg cada 8 semanas por vía subcutánea o placebo55. En la semana 22 el porcentaje de pacientes con respuesta clínica y remisión fue significativamente más alto en los tratados con ustekinumab que en los que recibieron placebo. En la actualidad están en marcha estudios en fase 3 de esta molécula para el tratamiento de la enfermedad de Crohn.
Por ahora, en el caso de que el ustekinumab desee indicarse en pacientes con enfermedad de Crohn, debe solicitarse su uso como «medicamento en situaciones especiales». La dosis recomendada para la inducción de la remisión de los pacientes con enfermedad de Crohn no está bien establecida, siendo la pauta más utilizada actualmente de 90mg por vía subcutánea en las semanas 0, 1, 2 y 3 y, posteriormente, tratamiento de mantenimiento 90mg por vía subcutánea cada 8 semanas.
Los efectos secundarios son escasos y no difieren del grupo placebo en los ensayos clínicos para enfermedad de Crohn. No obstante, los datos procedentes de pacientes con psoriasis muestran un excelente perfil de seguridad, siendo los efectos secundarios leves y poco frecuentes.
Otras interleucinasTanto la interleucina-13 como la interleucina-6 son citoquinas proinflamatorias que han sido probadas como potenciales dianas terapéuticas en la EII. Hasta la fecha, los resultados de los estudios con fármacos dirigidos contra estas moléculas han sido modestos56,57. En la actualidad se están evaluando nuevas moléculas contra estas citoquinas, para el tratamiento de la EII58.
Administración de citoquinas antiinflamatoriasLa administración de citoquinas antiinflamatorias podría ser un abordaje terapéutico en el tratamiento de la EII. Las citoquinas antiinflamatorias que han sido evaluadas como posibles tratamientos en los pacientes con EII son la interleucina-10, la interleucina-11 y el interferón-β59–66. Desafortunadamente, en los estudios llevados a cabo estas citoquinas no han demostrado ser útiles en el tratamiento de la EII.
Bloqueo de la adhesión leucocitariaEl etrolizumab es un anticuerpo monoclonal humanizado frente a la subunidad β7 de las integrinas α4β7 y αEβ7. El estudio EUCALYPTUS es un ensayo clínico fase 2, aleatorizado, doble ciego, controlado con placebo que evaluó la eficacia de etrolizumab en 124 pacientes con colitis ulcerosa moderada-grave que fueron aleatorizados a recibir etrolizumab 100mg en las semanas 0, 4 y 8 o etrolizumab 420mg (dosis de carga) y después 300mg en las semanas 2, 4 y 8 o placebo67. El porcentaje de pacientes que alcanzó la remisión clínica (Mayo≤2 con ningún subescore mayor de 1) fue del 21% en el grupo de etrolizumab 100mg, del 8% en el grupo de etrolizumab 300mg y del 0% en el grupo placebo (p=0,004 y p=0,05, respectivamente comparados con placebo). En la actualidad se están llevando a cabo estudios en fase 3 para confirmar estos prometedores resultados.
Por otro lado, las quimioquinas constituyen una familia compleja de proteínas recientemente identificadas como citoquinas con actividad quimiotáctica y activadora de diferentes tipos celulares del sistema inmune, que se diferencian de los quimioatrayentes clásicos, en su especificidad, por subtipos particulares de leucocitos. Estas moléculas están implicadas en el reclutamiento y migración de los leucocitos a la mucosa intestinal. Se ha descrito que podría existir una expresión aberrante del receptor 9 de las quimioquinas que se expresa en el intestino delgado y en el colon y podría ser una diana terapéutica en la enfermedad de Crohn68,69.
Vercirnón es un inhibidor oral del receptor de la quimoquina 9 que ha sido evaluado en un ensayo clínico doble ciego, controlado con placebo en pacientes con enfermedad de Crohn. Los pacientes fueron aleatorizados a recibir 250mg/24h, 250mg/12h o 500mg/24h o placebo durante 12 semanas. Los objetivos primarios fueron la respuesta en la semana 8 y 52. En la fase de inducción, únicamente el grupo que recibió 500mg/día presentó un porcentaje de respuesta significativamente más alta que placebo. En la semana 52, el 47% de los pacientes que recibieron vercirnón presentaron remisión en comparación con el 31% del grupo placebo70. En la actualidad se están llevando a cabo estudios fase 3 para evaluar la eficacia de esta molécula en la EII.
Bloqueo de las cascadas de señales mediadas por citoquinasInhibidores de la Janus KinasasLa implicación de las Janus kinasas (JK) JK1 y JK3 en el proceso de transducción de familia de los receptores de interleucina-2 e interleucina-6 (como la interleucina-12 y la interleucina-23) ha hecho que la inhibición de las JK sea una potencial diana terapéutica en la EII. El tofacitinib es un inhibidor de las JK 1, 2 y 3 capaz de modular las señales de un extenso grupo de citoquinas proinflamatorias, como las interleucinas-2, 4, 7, 9, 15 y 21. Estas integrinas participan en la activación, proliferación y funciones de los linfocitos.
En un ensayo clínico fase 2 aleatorizado, controlado con placebo se evaluó la eficacia de tofacitinib en pacientes con colitis ulcerosa moderada-grave67. Los pacientes fueron aleatorizados a recibir 4 dosis distintas de tofacitinib o placebo durante 8 semanas. En este estudio, tan solo el grupo que recibió la dosis más alta (15mg/12h) presentó una respuesta significativamente más alta que en el grupo placebo (78 vs. 42%). Con respecto a la tasa de pacientes con cicatrización mucosa, fue del 18% en el grupo que recibió 3mg, del 30% en el grupo de 10mg, del 27% en el grupo de 15mg y del 2% en el grupo placebo67. El perfil de seguridad con este fármaco fue bueno. En los pacientes que recibieron tofacitinib se observó un aumento en el colesterol LDL y HDL por un mecanismo no explicado, ya que teóricamente solo actúa sobre las JK que se expresan específicamente en las células del sistema inmune. En la actualidad se está llevando a cabo un estudio fase 3 para evaluar la eficacia y seguridad de tofacitinib en los pacientes con colitis ulcerosa.
LaquinimodEl laquinimod es una pequeña molécula sintética que, por un mecanismo no bien identificado, transforma las células T en un fenotipo antiinflamatorio disminuyendo las citoquinas proinflamatorias71. En un estudio en fase 2 a doble ciego, controlado con placebo de búsqueda de dosis los pacientes fueron aleatorizados a recibir laquinimod 0,5; 1; 1,5 o 2mg al día o placebo durante 8 semanas. Sorprendentemente, los mayores porcentajes de remisión y respuesta se obtuvieron en el grupo de pacientes tratados con la dosis más baja de laquinimod72.
MasitinibLos mastocitos del tracto gastrointestinal son centinelas del sistema inmune que se localizan en la mucosa y submucosa del tubo digestivo en la barrera entre el huésped y el ambiente. La activación y proliferación de los mastocitos puede observarse en ciertos procesos, incluida la EII. El masitinib es un inhibidor potente y selectivo de las tirosina kinasa que se dirige al receptor c-kit (expresado en los mastocitos), pero también modula otras citoquinas73. En la actualidad se está llevando a cabo un estudio en fase 2 en pacientes con enfermedad de Crohn.
ConclusionesEn los últimos tiempos se han incorporado nuevas moléculas al arsenal terapéutico de los pacientes con EII. El golimumab es un anticuerpo monoclonal que se dirige contra el TNF y que ha demostrado ser eficaz en el tratamiento de la colitis ulcerosa. Asimismo, ha sido aprobado en Europa el uso de CT-P13 (infliximab biosimilar) para las mismas indicaciones que el infliximab original, lo que supone disponer de una molécula dirigida contra el anti-TNF con la oportunidad de ahorro de costes asociados al tratamiento biológico. Más recientemente, vedolizumab, un anticuerpo monoclonal dirigido frente a las integrinas α4β7 ha sido aprobado para el tratamiento de la enfermedad de Crohn y de la colitis ulcerosa, con las ventajas de ser selectivo en el control de la inflamación en el tubo digestivo y de presentar un mecanismo de acción diferente a los clásicos anti-TNF.
Por otra parte, el estudio de la fisiopatología de la EII ha llevado a la identificación de numerosas dianas terapéuticas. En la actualidad se están desarrollando un gran número de moléculas, algunas de las cuales vendrán en un futuro a ampliar las opciones terapéuticas en los pacientes con EII.
Finalmente, en los próximos años los estudios deberán ir dirigidos a identificar factores predictores de respuesta a los distintos fármacos biológicos para la EII con el fin de seleccionar, de forma más personalizada, la mejor alternativa terapéutica para cada paciente.
Conflicto de interesesM. Chaparro: conferencias, soporte para investigación y/o actividades formativas: MSD, AbbVie, Hospira, Dr. Falk Pharma.
J.P. Gisbert: asesoramiento científico, conferencias, soporte para investigación y/o actividades formativas: MSD, AbbVie, Takeda, Hospira, Kern Pharma, Pfizer, Janssen, Ferring, Faes Farma, Shire Pharmaceuticals, Chiesi, Laboratorios Casen Fleet, Otsuka Pharmaceutical, Uriach y Dr. Falk Pharma y Gebro Pharma.

