




El manejo terapéutico de la esteatohepatitis no alcohólica se basa en la restitución de las alteraciones metabólicas, la pérdida de peso y los fármacos capaces de mejorar la esteatosis, la inflamación, la degeneración balonizante y la fibrosis. Las intervenciones sobre el estilo de vida basadas en dieta mediterránea e incremento de la actividad física configuran la primera línea del manejo de esta enfermedad. En pacientes en los que fracasa la intervención de estilo de vida se han de utilizar fármacos. Existen numerosos fármacos en desarrollo, entre los que destacan los agonistas FXR, los agonistas PPAR y los agonistas GLP-1R. Terapias dirigidas a las diferentes lesiones histológicas están también en desarrollo para mejorar la esteatosis, la inflamación, la degeneración balonizante, la apoptosis y la fibrosis. La cirugía bariátrica y la endoscopia terapéutica avanzada de la obesidad están reservadas a pacientes con obesidad mórbida en los que fracasan todas las opciones terapéuticas disponibles.
Management of non-alcoholic steatohepatitis is focused on restitution of metabolic derangement, weight loss and drugs able to improve steatosis, ballooning and fibrosis. Life-style interventions based on Mediterranean diet and increasing physical activity are the first line therapy. In patients with unsuccessful life-style intervention several drugs are under development: agonist PPAR, agonist GLP-1R and agonist FXR together with drugs focussing on inflammation, ballooning, apoptosis and fibrosis. Bariatric surgery or advanced endoscopy are reserved for morbid obese without response to life-style intervention and weighting loss drugs.
En los últimos años la importancia de la enfermedad por hígado graso no alcohólico (EHGNA) ha aumentado considerablemente, hasta el punto de que se ha situado como la enfermedad hepática más prevalente y una de las principales causas de trasplante hepático en el mundo occidental1. Se estima que la EHGNA se detecta en entre el 20 y el 30% de la población, aumenta paulatinamente con la edad y es más frecuente en hombres que en mujeres (estas diferencias son más acusadas en jóvenes menores de 50 años)2. Al tratarse de una enfermedad de muy alta prevalencia, son mucho más los pacientes con una enfermedad benigna con escaso impacto en su pronóstico, pero el pequeño porcentaje de pacientes con formas más avanzadas suponen la primera causa de enfermedad hepática en el mundo occidental. En pacientes con esteatohepatitis no alcohólica y, sobre todo, en aquellos que presentan fibrosis existe un riesgo aumentado de progresión de la enfermedad hepática, al mismo tiempo que aumenta el riesgo cardiovascular y de neoplasias sólidas. Por todo ello, en los pacientes con riesgo de progresión de la enfermedad (esteatohepatitis, fibrosis, edad >60años, género masculino o presencia de diabetes mellitus tipo2) está indicado el tratamiento. La EHGNA presenta 4 fenotipos asociados: la obesidad, la diabetes, el síndrome metabólico y la EHGNA en pacientes no obesos metabólicamente sanos. De forma muy resumida y con un constante cruce de vías patogénicas, la EHGNA consiste en la puesta en marcha de un elevado aporte de ácidos grasos libres al hígado, especialmente acusado en la obesidad por la resistencia a la insulina (tanto hepática como muscular y adiposa), por otro lado, un incremento de la lipogénesis de novo, sobre todo debido a la hiperinsulinemia, estado característico del síndrome metabólico y la diabetes mellitus tipo2, de forma que la secreción de triglicéridos en forma de VLDL no es suficiente y se produce la acumulación de grasa. La lipotoxicidad es responsable del estrés oxidativo con aumento de la producción de radicales libres de oxígeno y el estrés del retículo endoplásmico que condicionan la puesta en marcha de mecanismos de apoptosis, alteración de la respuesta inmune y activación de la célula hepática estrellada como catalizadora de la progresión de la fibrosis. Estos fenómenos patogénicos tienen lugar en el contexto de la interacción del exposoma, el microbioma y el genoma del paciente. La microbiota intestinal puede afectar al hígado por varios mecanismos: a) alteración de la barrera intestinal, lo que facilita la translocación bacteriana y la llegada de productos bacterianos como los lipopolisacáridos; b) alteración del metabolismo de los ácidos grasos de cadena corta y la producción intestinal de alcohol; c) alteración del metabolismo de los ácidos biliares, de forma que la microbiota intestinal puede inhibir la síntesis de ácidos biliares primarios en el hígado mediante la inhibición del receptor farnesoideX (FXR)3.
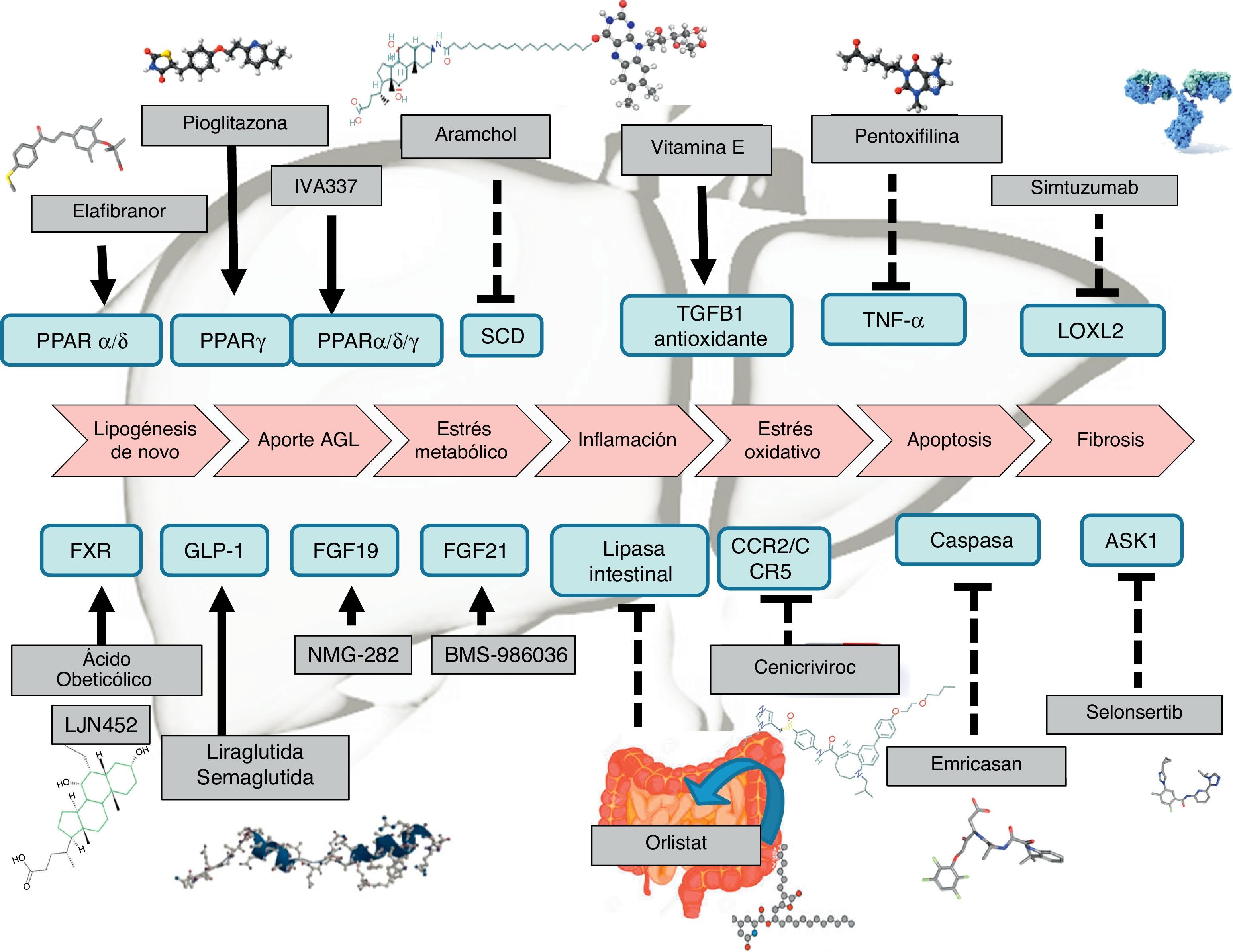
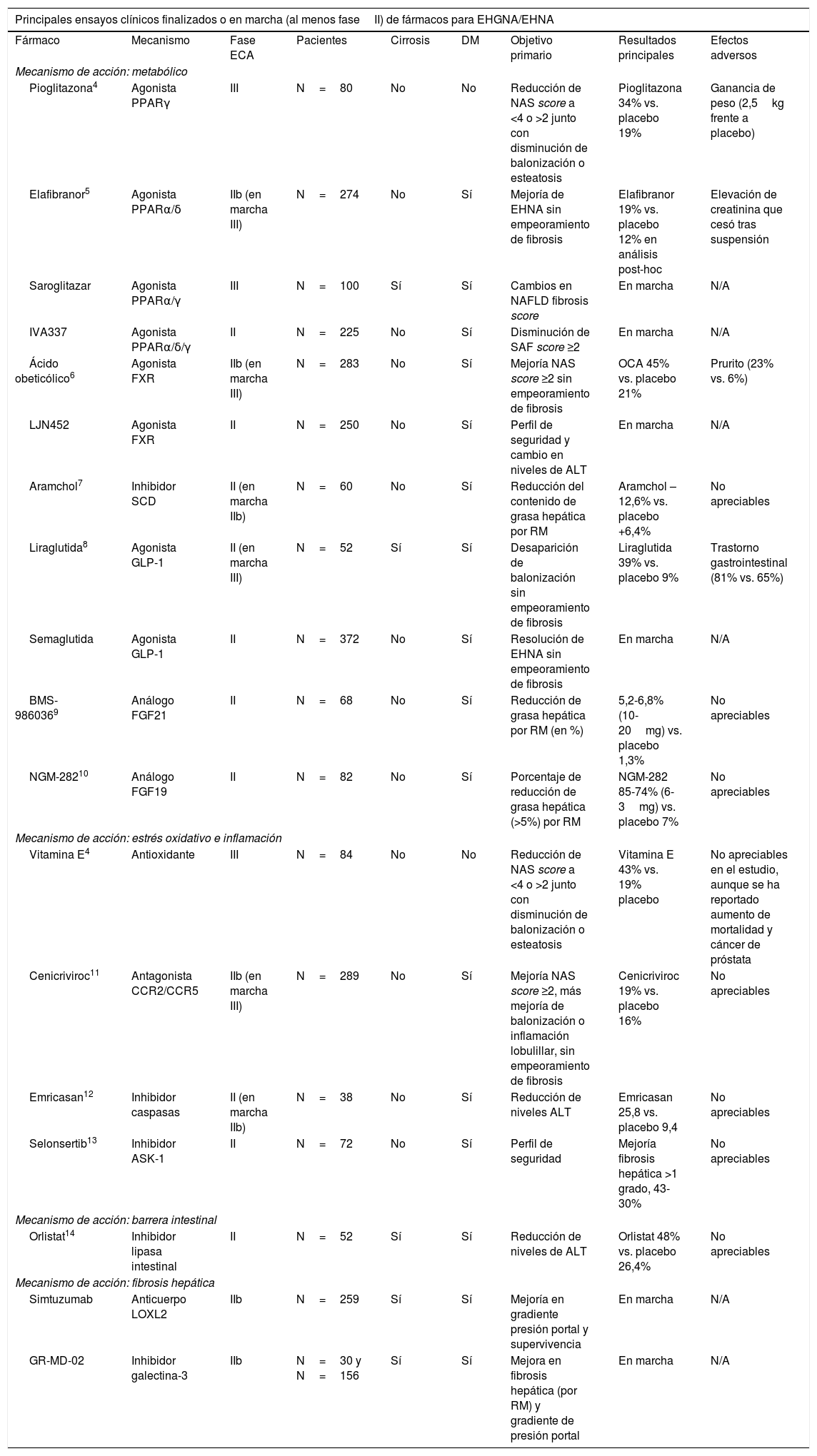
Las recomendaciones terapéuticas en pacientes en los que fracasan las intervenciones sobre el estilo de vida se han diseñado para abordar la obesidad, la diabetes o los trastornos metabólicos del paciente. Así también, se están diseñando fármacos dirigidos a resolver directamente la esteatosis, la esteatohepatitis (inflamación y degeneración balonizante) y la fibrosis (tabla 1). Los fármacos que han mostrado efectos beneficiosos son la pioglitazona y la vitaminaE, pero tanto la FDA como la EMA no recomiendan ningún tratamiento farmacológico en el momento actual y las guías AASLD-EASL-EASD recomiendan su uso solo en pacientes con diagnóstico histológico de esteatohepatitis no alcohólica, no diabéticos y no cirróticos15.
Principales ensayos clínicos aleatorizados y controlados en pacientes con EHGNA/EHNA
| Principales ensayos clínicos finalizados o en marcha (al menos faseII) de fármacos para EHGNA/EHNA | ||||||||
|---|---|---|---|---|---|---|---|---|
| Fármaco | Mecanismo | Fase ECA | Pacientes | Cirrosis | DM | Objetivo primario | Resultados principales | Efectos adversos |
| Mecanismo de acción: metabólico | ||||||||
| Pioglitazona4 | Agonista PPARγ | III | N=80 | No | No | Reducción de NAS score a <4 o >2 junto con disminución de balonización o esteatosis | Pioglitazona 34% vs. placebo 19% | Ganancia de peso (2,5kg frente a placebo) |
| Elafibranor5 | Agonista PPARα/δ | IIb (en marcha III) | N=274 | No | Sí | Mejoría de EHNA sin empeoramiento de fibrosis | Elafibranor 19% vs. placebo 12% en análisis post-hoc | Elevación de creatinina que cesó tras suspensión |
| Saroglitazar | Agonista PPARα/γ | III | N=100 | Sí | Sí | Cambios en NAFLD fibrosis score | En marcha | N/A |
| IVA337 | Agonista PPARα/δ/γ | II | N=225 | No | Sí | Disminución de SAF score ≥2 | En marcha | N/A |
| Ácido obeticólico6 | Agonista FXR | IIb (en marcha III) | N=283 | No | Sí | Mejoría NAS score ≥2 sin empeoramiento de fibrosis | OCA 45% vs. placebo 21% | Prurito (23% vs. 6%) |
| LJN452 | Agonista FXR | II | N=250 | No | Sí | Perfil de seguridad y cambio en niveles de ALT | En marcha | N/A |
| Aramchol7 | Inhibidor SCD | II (en marcha IIb) | N=60 | No | Sí | Reducción del contenido de grasa hepática por RM | Aramchol –12,6% vs. placebo +6,4% | No apreciables |
| Liraglutida8 | Agonista GLP-1 | II (en marcha III) | N=52 | Sí | Sí | Desaparición de balonización sin empeoramiento de fibrosis | Liraglutida 39% vs. placebo 9% | Trastorno gastrointestinal (81% vs. 65%) |
| Semaglutida | Agonista GLP-1 | II | N=372 | No | Sí | Resolución de EHNA sin empeoramiento de fibrosis | En marcha | N/A |
| BMS-9860369 | Análogo FGF21 | II | N=68 | No | Sí | Reducción de grasa hepática por RM (en %) | 5,2-6,8% (10-20mg) vs. placebo 1,3% | No apreciables |
| NGM-28210 | Análogo FGF19 | II | N=82 | No | Sí | Porcentaje de reducción de grasa hepática (>5%) por RM | NGM-282 85-74% (6-3mg) vs. placebo 7% | No apreciables |
| Mecanismo de acción: estrés oxidativo e inflamación | ||||||||
| Vitamina E4 | Antioxidante | III | N=84 | No | No | Reducción de NAS score a <4 o >2 junto con disminución de balonización o esteatosis | Vitamina E 43% vs. 19% placebo | No apreciables en el estudio, aunque se ha reportado aumento de mortalidad y cáncer de próstata |
| Cenicriviroc11 | Antagonista CCR2/CCR5 | IIb (en marcha III) | N=289 | No | Sí | Mejoría NAS score ≥2, más mejoría de balonización o inflamación lobulillar, sin empeoramiento de fibrosis | Cenicriviroc 19% vs. placebo 16% | No apreciables |
| Emricasan12 | Inhibidor caspasas | II (en marcha IIb) | N=38 | No | Sí | Reducción de niveles ALT | Emricasan 25,8 vs. placebo 9,4 | No apreciables |
| Selonsertib13 | Inhibidor ASK-1 | II | N=72 | No | Sí | Perfil de seguridad | Mejoría fibrosis hepática >1 grado, 43-30% | No apreciables |
| Mecanismo de acción: barrera intestinal | ||||||||
| Orlistat14 | Inhibidor lipasa intestinal | II | N=52 | Sí | Sí | Reducción de niveles de ALT | Orlistat 48% vs. placebo 26,4% | No apreciables |
| Mecanismo de acción: fibrosis hepática | ||||||||
| Simtuzumab | Anticuerpo LOXL2 | IIb | N=259 | Sí | Sí | Mejoría en gradiente presión portal y supervivencia | En marcha | N/A |
| GR-MD-02 | Inhibidor galectina-3 | IIb | N=30 y N=156 | Sí | Sí | Mejora en fibrosis hepática (por RM) y gradiente de presión portal | En marcha | N/A |
Por último, el interés por esta enfermedad es creciente, como demuestran las numerosas revisiones publicadas en los últimos meses sobre el tratamiento farmacológico y no farmacológico de la esteatohepatitis no alcohólica16-21.
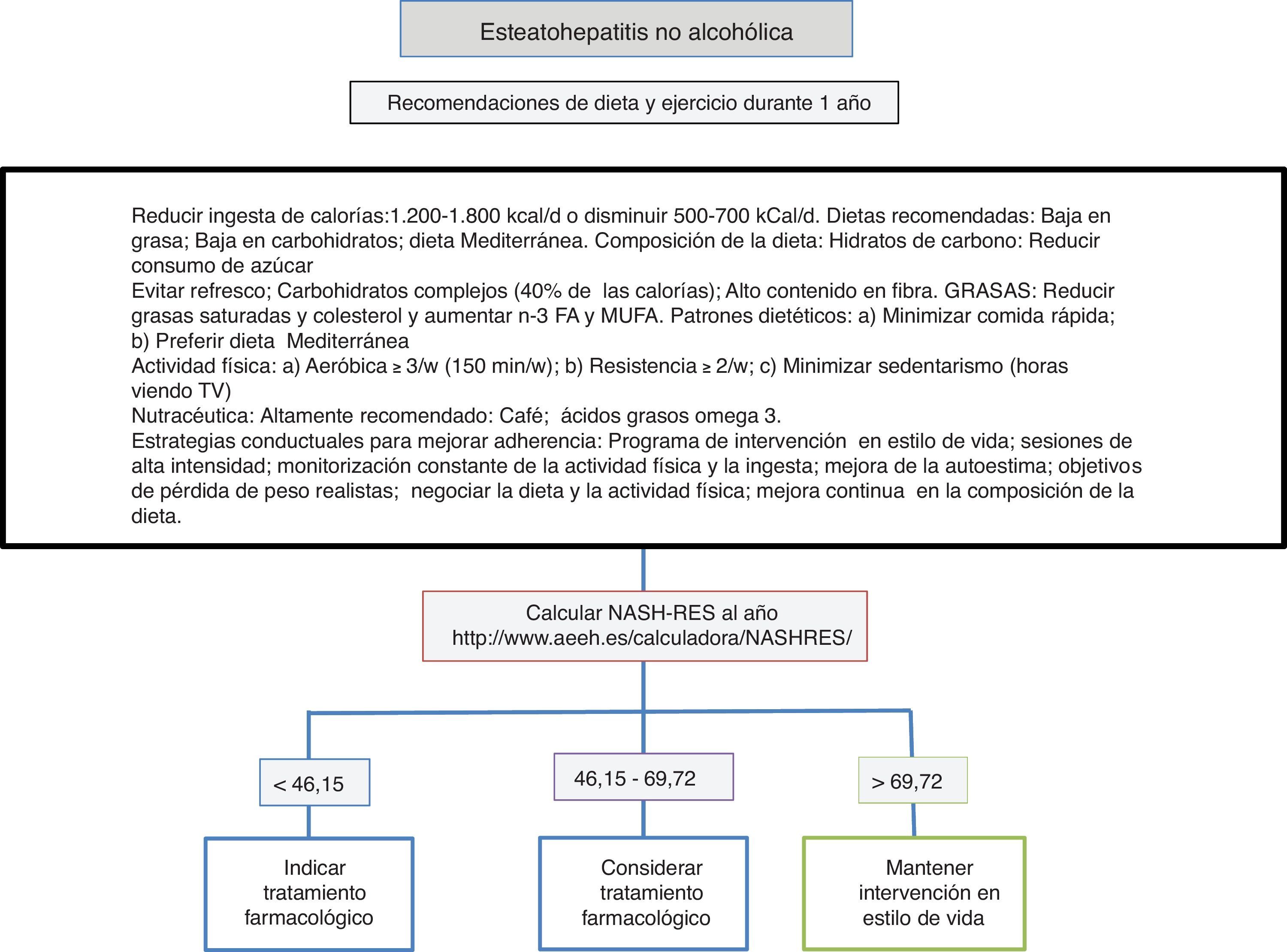
Dieta, actividad física y ejercicio en el manejo de la esteatohepatitis no alcohólicaLa primera línea consiste en una intervención sobre el estilo de vida promoviendo una dieta mediterránea, un abandono del sedentarismo e incremento de la actividad física y realización de ejercicio físico aeróbico moderado con el objetivo de inducir pérdida de peso que garantice la mejoría de la enfermedad hepática (fig. 1). La dieta hipocalórica promueve pérdida de peso, pero no se han detectado diferencias entre la dieta hipocalórica baja en grasas o baja en hidratos de carbono. Además, en pacientes sometidos a dietas isocalóricas, la ingesta de ácidos grasos monoinsaturados (como el aceite de oliva) o poliinsaturados (como el aceite de girasol) promueve la eliminación de la acumulación de grasa en el hígado (valorada mediante resonancia magnética espectroscópica). La dieta mediterránea rica en aceite de oliva, fibra, frutos secos, aceite de pescado rico en omega-3, frutas y verduras y con escaso consumo de azúcares refinados, grasas saturadas y carnes rojas procesadas previene el desarrollo de esteatosis hepática y esteatohepatitis22. Por otro lado, una dieta hipocalórica con un descenso de 500kCal/d baja en grasas (<30%) ha demostrado inducir resolución de la esteatohepatitis y regresión de la fibrosis en pacientes con pérdida de peso, de forma que el porcentaje de pacientes que mejoran es directamente proporcional al porcentaje de peso perdido. El sedentarismo (valorado como el número de horas que pasa el paciente viendo televisión sin interrupciones) y la actividad física espontánea (valorada como inactivo, mínimamente activo o actividad moderada) deben valorarse adecuadamente en cada paciente para prescribir el ejercicio físico idóneo (definido como una actividad realizada de forma premeditada en forma de ejercicio físico aeróbico, de resistencia, de alta intensidad intermitente o ejercicio vigoroso aeróbico) y la dieta más óptima. Los elementos principales consisten en evitar el sedentarismo de forma prolongada, por lo que se recomiendan las interrupciones, al menos cada hora, evitar la inactividad física y promover ejercicio físico aeróbico moderado con episodios de alta intensidad23. No se han encontrado diferencias entre el ejercicio físico aeróbico moderado y el ejercicio de resistencia24. En un estudio prospectivo con 293 pacientes a quienes se diagnosticó esteatohepatitis no alcohólica por biopsia se indicó una intervención mediante dieta hipocalórica baja en grasas, actividad física de al menos 200min a la semana, cuestionario de actividad y sesiones de reforzamiento de adherencia a la dieta en grupos conductuales. En los pacientes con pérdida de peso ≥10% la tasa de resolución de la esteatohepatitis no alcohólica fue del 90% y la regresión de al menos un estadio de fibrosis, del 81%25. En un análisis posterior se comprobó que en 261 pacientes con doble biopsia hepática, la normalización de transaminasas (<19 en mujeres y <30 en hombres), así como el porcentaje de pérdida de peso, la edad, la presencia basal de diabetes y un NAS score >5 permitían calcular las posibilidades de resolución de la esteatohepatitis (NASH-RES)26. Los pacientes con un resultado inferior a 46,12 deben ser considerados para tratamiento farmacológico, ya que la posibilidad de resolver la enfermedad con esta estrategia es casi nula. En cambio, aquellos con resultados superiores a 69,75 deben mantener la misma actitud en el tiempo y considerarse libres de riesgo de progresión de la enfermedad.
Terapia farmacológica mediante intervención en el estilo de vida e indicación de tratamiento farmacológico en pacientes sin opciones de resolución de la EHNA.")
El desarrollo de terapias contra la EHGNA es un campo amplio de investigación actualmente. Existen medicamentos que tienen un desarrollo más avanzado y que por tanto están más cerca de la comercialización. Sin embargo, muchas otras terapias muestran resultados preliminares o tienen en marcha ensayos clínicos faseII o faseIII (tabla 1). A grandes rasgos, los fármacos pueden clasificarse según el mecanismo de acción: a) metabolismo, como pueden ser agonistas de los receptores peroxisome proliferator-activated receptor (PPAR)4, FXR6, fibroblast growth factor (FGF)9,10 y glucagon-like peptide-1 (GLP-1)8; b) estrés oxidativo e inflamación, como la vitaminaE4, cenicriviroc27, emricasan12 y selonsertib13; c) fibrosis hepática, como simtuzumab o GR-MD-02. Estos últimos estarían indicados en pacientes con cirrosis hepática; de hecho, tienen en marcha ensayos clínicos en los que se evalúa el beneficio sobre el gradiente de presión portal y la supervivencia.
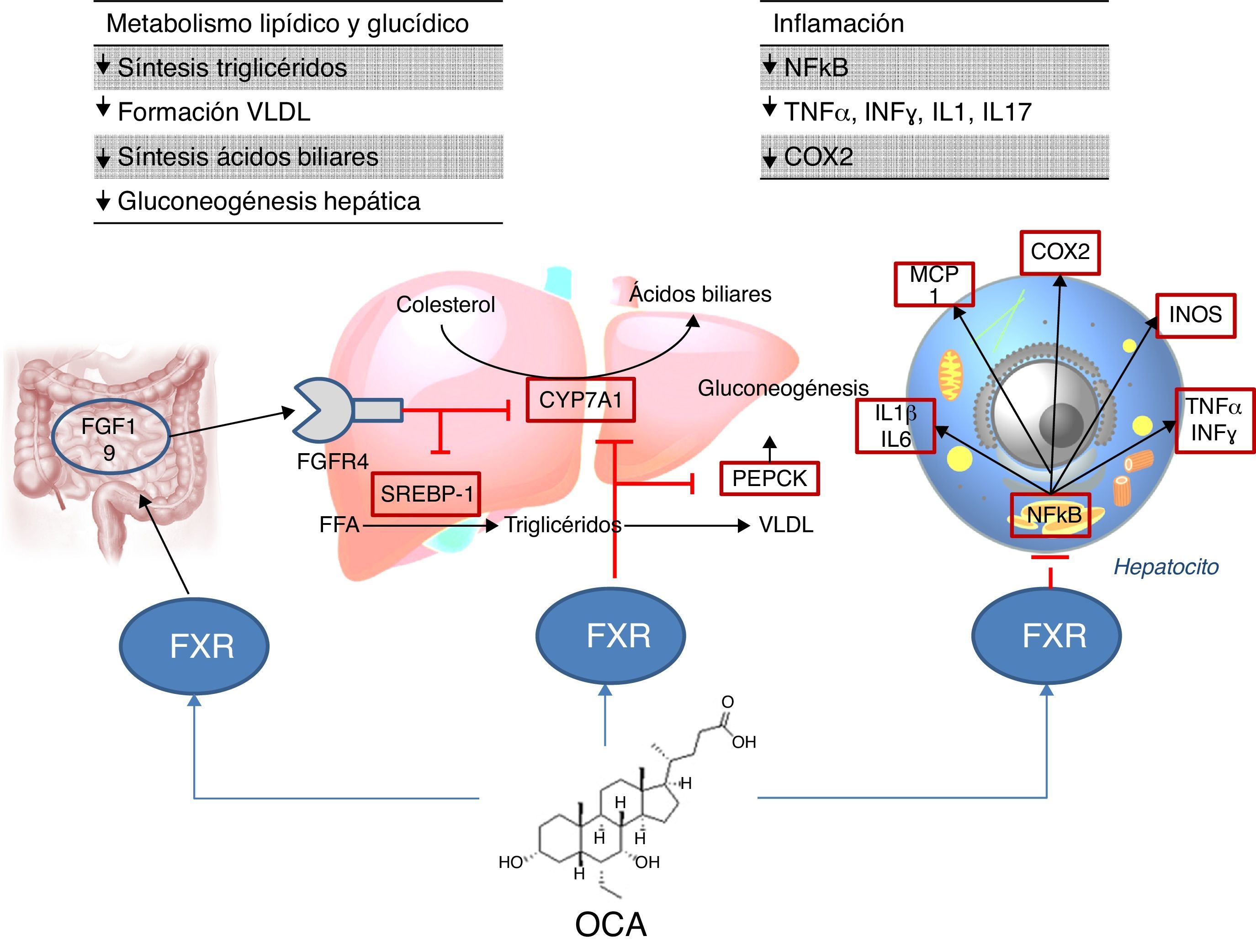
Lugar de acción: metabolismoAgonistas FXRLa activación de FXR regula el metabolismo del colesterol (fig. 2)28. Una vez se forman los ácidos biliares a partir del colesterol, estos son almacenados en la vesícula biliar desde donde se segregan al intestino delgado tras la ingesta de alimentos, reciclándose el 95% de los ácidos biliares mediante la circulación enterohepática. Para compensar la pérdida del 5% de ácidos biliares excluidos de la circulación enterohepática, el hígado los sintetiza de nuevo pudiendo provocar toxicidad celular por su acumulación. Para evitar esto, se unen al FXR suprimiendo la expresión de la colesterol-7α-hidroxilasa (CYP7A1) reduciendo la nueva síntesis. Por otro lado, el FXR induce la expresión de FGF15/19 en los enterocitos activando la ruta de señalización JNK (que también disminuye la expresión génica de CYP7A1)29. En cuanto al metabolismo glucídico y lipídico, se ha demostrado que el FXR juega un papel importante a través de la regulación de la expresión del gen de la fosfoenolpiruvato carboxiquinasa (PEPCK) y de otros genes implicados en el metabolismo de las lipoproteínas30. Además, diversos estudios en ratones knock-out para FXR (FXR–/–) han objetivado que: a) presentaban niveles incrementados de glucosa y ácidos grasos libres circulantes (desarrollando resistencia a la insulina y esteatosis hepática)31; b) aumentaban los niveles de HDL (reduciéndose la expresión de genes implicados en el transporte inverso del colesterol) y colesterol no HDL, así como la síntesis de apolipoproteínaB y la absorción intestinal de colesterol30; c) descendían los niveles séricos de triglicéridos al disminuir la expresión hepática de SREBP-1c32. Por otro lado, FXR ejerce una acción antiinflamatoria hepática mediante la supresión de factor nuclear-κB (NFκB)33. A su vez, su activación reduce la expresión de citoquinas proinflamatorias en los macrófagos (inducida por lipopolisacáridos), lo que sugiere que reduce directamente la respuesta inflamatoria en las células inmunes34.

El ácido obeticólico (OCA) (6-etilquenodesoxicólico) es un derivado semisintético del ácido quenodesoxicólico, con mayor potencia que este35, que ejerce una acción agonista de FXR. El perfil farmacocinético de OCA ha sido bien establecido. Se conjuga con glicina o taurina en el hígado siendo secretado posteriormente en la bilis. Estos conjugados: a) se absorben en el intestino delgado, entrando en la circulación enterohepática; b) son deconjugados en íleon y colon por la microbiota intestinal; c) liberan el ácido, que puede ser reabsorbido o excretado en las heces, la ruta principal de eliminación (<3% se excreta en orina). Tanto el OCA como sus conjugados circulan en su mayoría (>99%) unidos a proteínas plasmáticas. Según la Agencia Europea de Medicamentos36, la concentración plasmática máxima del fármaco se alcanza a las 2h, no alterándose por la administración conjunta con alimentos. Dado que el OCA se metaboliza en el hígado, su nivel plasmático se incrementa en pacientes con insuficiencia hepática moderada y grave, recomendando disminuir la dosis en estos casos. En individuos con insuficiencia renal (filtrado glomerular hasta 50ml/min/1,73m2) no se ha demostrado un impacto negativo, aunque se desconocen sus efectos en presencia de insuficiencia renal avanzada.
El estudio FLINT analizó el papel del OCA en la EHGNA6; se trata de un ensayo clínico aleatorizado, multicéntrico, doble ciego, controlado con placebo y en grupos paralelos, que incluyó pacientes con esteatohepatitis no alcohólica en estadio no cirrótico. El objetivo fue evaluar el tratamiento con OCA oral (25mg al día) durante 72 semanas. Se observó un 45% de mejoría histológica (definida como mejoría ≥2 puntos en NAS score sin empeoramiento de la fibrosis) en pacientes con OCA en comparación con el 21% de pacientes del grupo placebo. Asimismo, se observó mejoría en los pacientes tratados en términos de esteatosis hepática, inflamación, balonización hepatocitaria y fibrosis, así como una disminución en los niveles de transaminasas. Sin embargo, la proporción de pacientes con resolución de esteatohepatitis no fue significativamente distinta entre ambos grupos. En otro ensayo clínico faseII, se evaluó el impacto del OCA sobre la sensibilidad a la insulina en pacientes con EHGNA y diabetes mellitus tipo2, evidenciándose una mejoría del 24% en pacientes tratados frente a un empeoramiento del 5% en aquellos que recibieron placebo37.
En cuanto a efectos adversos, los pacientes con OCA presentaron un aumento del colesterol total y LDL (0,38mmol/l [IC95%: 0,16-0,60] p=0,0009; 0,45mmol/l [IC95%: 0,26-0,65] p=0,0001, respectivamente) y una disminución del HDL (–0,06mmol/l [IC95%: –0,10-0,01] p=0,01). Si este perfil aterogénico supone un incremento de riesgo cardiovascular es algo que debe confirmarse en nuevos estudios. Por otro lado, el fármaco fue bien tolerado aunque el porcentaje de prurito fue superior en OCA que en placebo (23% vs. 6%, p<0,0001)6 (también observado en colangitis biliar primaria38), siendo dosis dependiente39. Tras la retirada del fármaco, la alteración del perfil lipídico regresó a niveles pretratamiento y el prurito desapareció. La función renal ha sido evaluada en un análisis post-hoc del estudio FLINT40, tanto en aquellos con OCA como con placebo. Se ha observado una disminución del filtrado glomerular en los pacientes tratados con OCA con respecto al grupo placebo (–1,9±7,8ml/min/año frente a –0,2±7,2ml/min/año; p=0,02), por lo que se recomienda la monitorización de la función renal en pacientes con esteatohepatitis tratados con OCA.
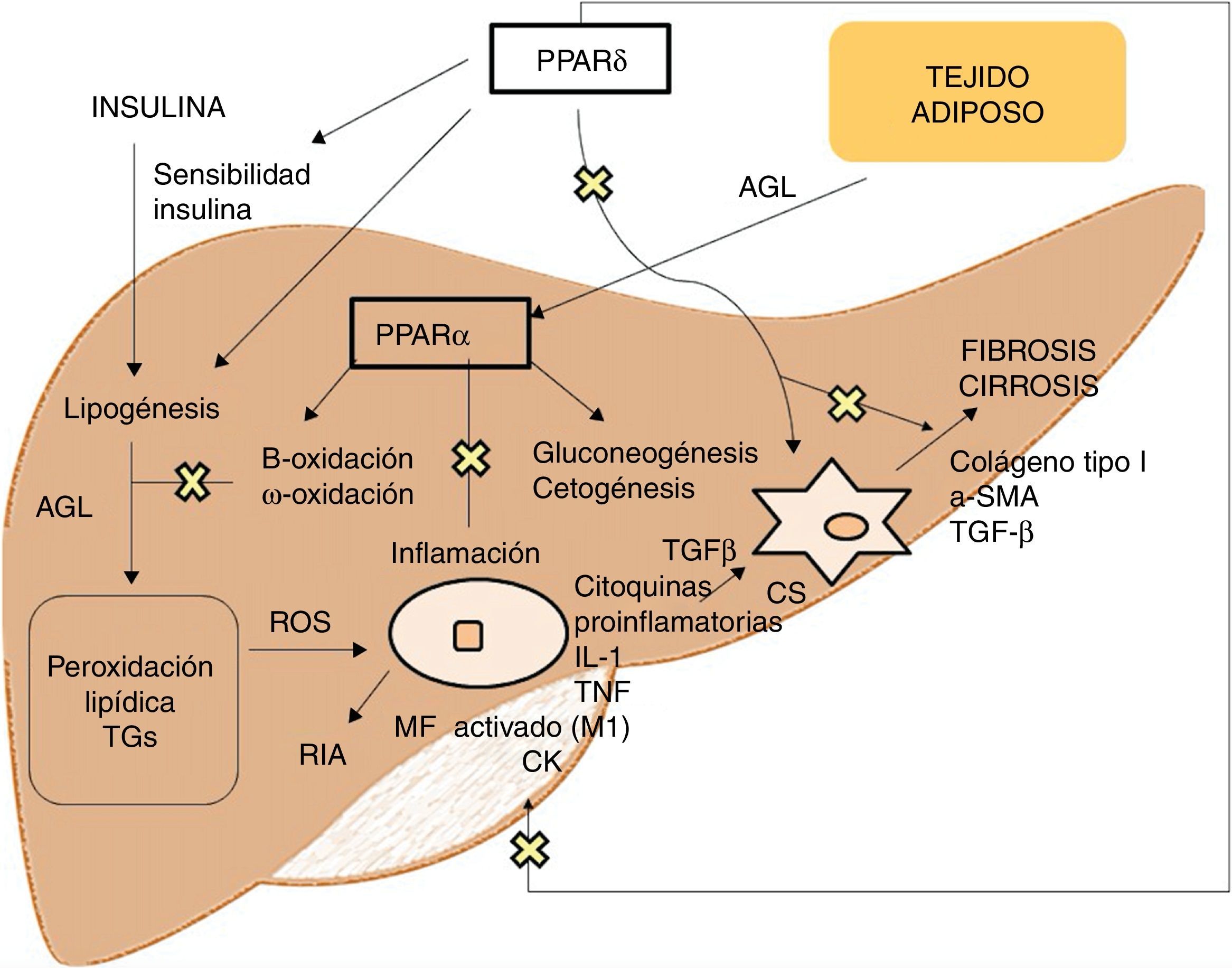
Agonistas PPARLos receptores PPAR actúan como factores de transcripción y regulan la homeostasis energética, la respuesta inflamatoria y el metabolismo lipídico y glucídico41 (fig. 3). Basados en su homología, se han identificado 3 isotipos. Los PPARα, localizados en el cromosoma 22q12-13.1, se expresan fundamentalmente en el hígado y el tejido adiposo marrón, así como en macrófagos y células parenquimatosas42. Los PPARδ, localizados en el cromosoma 6p21.2-21.1, tienen una expresión muy ubicua en diferentes tejidos43. Por último, los PPARγ, localizados en el cromosoma 3p25, son ampliamente expresados en el tejido adiposo con baja expresión en hígado44.

La activación de PPARα consigue la regulación de la respuesta inflamatoria a través de la modulación de IL-6, lo cual se traduce en una supresión de las proteínas de respuesta de fase aguda estimuladas por esta, como son el amiloide séricoA, la α2-macroglobulina y el plasminógeno45, así como su efecto regulador en la expresión de genes antiinflamatorios como IL-1ra y IkBα, un inhibidor citoplasmático de NFkB que juega un papel fundamental en la respuesta inmune e inflamatoria46. La activación de PPARδ mejora la sensibilidad a la insulina, incrementa el transporte y oxidación de ácidos grasos, disminuye la lipogénesis y tiene actividad antiinflamatoria en macrófagos y células de Kupffer47. El beneficio de PPARδ en la homeostasis de la glucosa se basa en la inducción de moléculas de señalización en el tejido adiposo que indirectamente provocan una mejoría en el músculo de la eliminación de la glucosa, como son por un lado el TNFα, plasminógeno y leptina, que interaccionan con las vías de señalización de la insulina, y por otro los ácidos grasos libres (AGL), los cuales determinan la sensibilidad a la insulina muscular. Los agonistas de este receptor promueven la expresión de genes implicados en el metabolismo de AGL así como la captación de estos por el adipocito, disminuyendo su disponibilidad en el músculo y mejorando la sensibilidad a la insulina muscular48. Además, en el tejido adiposo blanco, estimula la expresión de genes involucrados en el consumo y almacenamiento de ácidos grasos, incluyendo la lipoproteinlipasa, CD36/FAT, aP2 (proteína de unión adipocitaria a ácido graso) o carboxiquinasa fosfoenolpiruvato. De esta forma, su activación genera secuestro de lípidos en el tejido adiposo blanco para almacenaje o en el hígado para su oxidación, reduciendo su acumulación en el músculo esquelético lo que podrían interferir con la acción de la insulina49. Respecto al metabolismo lipídico, promueve el eflujo de colesterol por inducción de ABCA1 (transportador inverso de colesterol), inhibe la absorción intestinal de colesterol mediante la regulación negativa del gen Niemann-Pick C1-like, estimula la oxidación y uso de ácidos grasos aumentando la expresión de los genes diana que controlan estas vías metabólicas y regula la lipogénesis a través de la inducción de la expresión del gen inducido por insulina (Insig-1) que conlleva la supresión de enzimas clave en la biosíntesis de ácidos grasos, como SREBP-150. Además, promueve la diferenciación de los adipocitos y la redistribución de grasas del hígado y músculo al tejido adiposo, evitando el acúmulo intracelular de ácidos grasos y sus metabolitos (ácidos grasos-CoA, diacilglicerol y ceramidas). En macrófagos, atenúa la producción de citoquinas proinflamatorias (IL-1β, MIP-1α e ICAM-1) a través de vías de señalización dependientes de ERK1/2 y AKT/FoxO51 y regula la activación de macrófagos y células de Kupffer. En este sentido, PPARδ juega un papel clave en el balance entre macrófagos hepáticos M1, con potencial proinflamatorio y M2, con potencial antiinflamatorio, de manera que la activación del receptor promueve la polarización hacia fenotipo M2, resultando en una actividad antiinflamatoria más pronunciada. Un mecanismo bien conocido por el que controla la respuesta inflamatoria es mediante la interferencia negativa con vías de señalización proinflamatoria como son AP-1, NF-kB o STAT-3 en macrófagos M1 activados52. Por otra parte, PPARδ reduce y previene la fibrosis hepática ya que interviene en la activación, alteración fenotípica y mantenimiento en fase quiescente de las células estrelladas hepáticas, suprimiendo además la producción de colágeno tipoI, α-SMA y TGF-β. Este receptor puede bloquear las vías de señalización de TGF-β y la actividad promotora dependiente SMAD, antagonizando la activación y/o función de Smad3 en fibroblastos53.
Elafibranor (2-[2,6-dimetil-4-[3-[4-(metiltio) fenil]-3-oxo-1(E)-propenil]fenoxil]-2-ácido metilpropanoico), con su metabolito activo (GFT1007), es un agonista dual de los receptores PPARα y PPARδ, con mayor actividad PPARα (EC50: 45nmol/l para GFT505 y 15nmol/l para GFT1007) que PPARδ (EC50: 175 y 75nmol/l, respectivamente), pero sin actividad en PPARγ54. Se excreta fundamentalmente por vía biliar, siendo reabsorbido gran parte en la circulación enterohepática55. Elafibranor ha sido evaluado en EHGNA en diversos estudios desde 2011. Cariou et al.5 publicaron dos estudios en pacientes obesos (con dislipidemia o prediabetes, respectivamente) evidenciando por primera vez su efecto beneficioso. En ambos estudios el fármaco redujo la concentración de triglicéridos plasmáticos e incrementó HDL, mejoró la sensibilidad a la insulina (descenso de HOMA-IR) así como los marcadores de función hepática. No hubo eventos adversos importantes que limitaran el estudio, si bien se produjo un aumento moderado de las cifras de creatinina reversible tras la suspensión del fármaco. En 2013, Cariou et al.54 realizaron un ensayo clínico en pacientes obesos insulinorresistentes en los que se inducía un estado de hiperinsulinemia-euglucemia mediante la infusión de insulina y glucosa marcada con un isótopo no radiactivo. Comparado con placebo, elafibranor demostró mejorar la sensibilidad a insulina en hígado, músculo y tejido adiposo, además de mejorar también los niveles de transaminasas. En 2016, Ratziu et al.56 realizaron un ensayo clínico faseIIb donde se aleatorizaron pacientes con esteatohepatitis no cirróticos a recibir elafibranor 80mg, 120mg o placebo durante un año. La dosis de 120mg fue superior a placebo, tanto en la resolución histológica de esteatohepatitis sin empeorar la fibrosis como en la mejoría de NAS score. La mejoría histológica se acompañó de un beneficio en los marcadores de función hepática y métodos no invasivos de fibrosis. Nuevamente, el fármaco fue bien tolerado aunque se observó un ligero incremento de las cifras de creatinina (reversible tras la suspensión del medicamento).
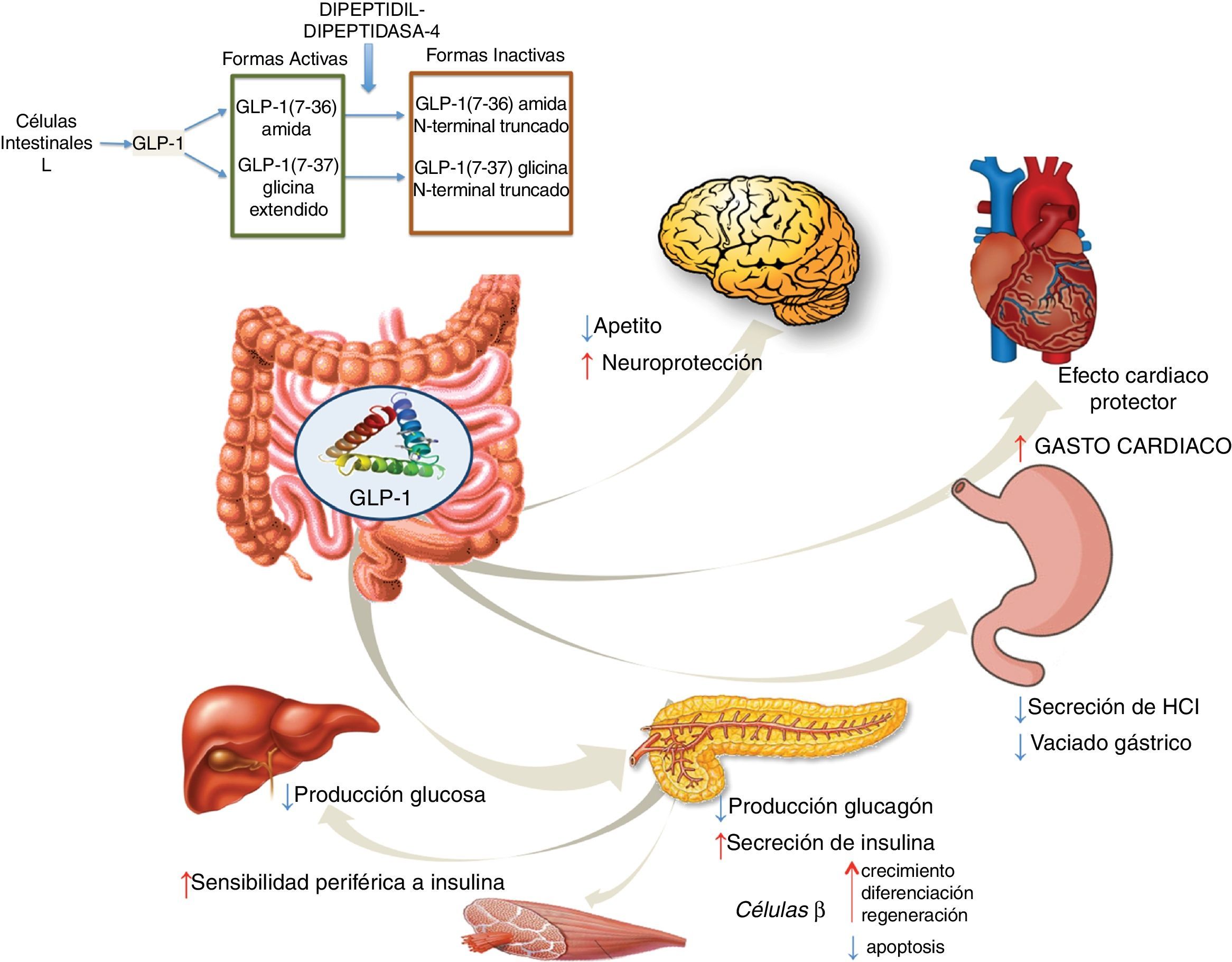
Agonistas GLP-1RGLP-1 es una hormona intestinal con carácter de incretina que se secreta tanto en personas sanas como en pacientes diabéticos tipo2, tras la ingestión de alimentos ricos en azúcares y grasas57. GLP-1 provoca una respuesta pancreática incrementando la síntesis de insulina e inhibiendo la de glucagón. GLP-1 ha demostrado efectos sobre diferentes dianas: a) sobre la célula beta pancreática, promoviendo su crecimiento, diferenciación y regeneración, al tiempo que inhibe la apoptosis; b) en el estómago reduce la secreción ácida y enlentece su vaciamiento58; c) en el cerebro disminuye el apetito y aumenta la neuroprotección; d) disminuye la producción hepática de glucosa; e) mejora la sensibilidad periférica a la insulina; f) muestra efectos cardioprotectores aumentando el gasto cardíaco (fig. 4). Los análogos de GLP-1 han logrado mostrar reducción en los valores de enzimas hepáticas y del estrés oxidativo mejorándose el patrón histológico. A su vez se ha demostrado una acción directa celular hepática disminuyendo la lipogénesis e incrementando la oxidación de ácidos grasos59.

Liraglutida es un análogo GLP-1 de duración larga que fue aprobado para el control glucémico en pacientes con sobrepeso y diabetes mellitus tipo2. Los péptidos GLP-1 endógenos son degradados en pocos minutos por la enzima dipeptidil peptidasa-4 (DPP-4), mientras que liraglutida se metaboliza de forma mucho más lenta. El estudio LEAN (Liraglutide Efficacy and Action in NASH)8 es un ensayo multicéntrico, aleatorizado, doble ciego en el que se evaluó el efecto a 48 semanas de liraglutida frente a placebo en pacientes con esteatohepatitis no alcohólica definida como esteatosis macrovesicular (>5%) más balonización, presencia de hialina de Mallory por inmunohistoquímica e inflamación lobulillar, junto con IMC>25kg/m2. Pacientes con diabetes tipo2 fueron escogidos si mantenían estables controles glucémicos (hemoglobina glicada <9) y eran tratados con dieta o con dosis estable de metformina o sulfonilurea. El objetivo primario del estudio fue la mejoría histológica, definida como la resolución de esteatohepatitis sin empeoramiento de la fibrosis. Se reclutaron un total de 52 pacientes que se aleatorizaron en dos grupos, recibiendo cada uno de ellos liraglutida y placebo, respectivamente. Finalmente, 23 pacientes del grupo del fármaco estudiado y 22 del grupo placebo tenían biopsias al inicio y a las 48 semanas de comenzar el tratamiento. Nueve (39%) de los 23 pacientes del grupo tratado con liraglutida tuvieron resolución completa de la esteatohepatitis sin empeoramiento de la fibrosis, en comparación con 2 (9%) de 22 pacientes del grupo placebo. A su vez, 3 (38%) de 8 pacientes con diabetes tipo2 y 6 (40%) de 15 pacientes sin diabetes tipo2 alcanzaron el objetivo primario con liraglutida. Ninguno de los 2 pacientes del grupo placebo que obtuvieron mejoría histológica tenía diabetes tipo2. Además, como objetivos secundarios se obtuvieron datos de mejoría en el peso y cifras de glucemia con liraglutida que podrían tener un efecto favorable en el riesgo de desarrollar enfermedad cardiovascular y muerte prematura. Sin embargo, hacen falta más estudios con resultados a largo plazo para confirmar esta hipótesis. En cuanto a efectos adversos, liraglutida fue seguro y bien tolerado independientemente de la severidad de la enfermedad de base.
Inhibidores de la DPP-4Los inhibidores de la enzima DPP-4 encargada de la degradación del GLP-1 endógeno, tales como la sitagliptina y la vildagliptina, consiguen prolongar la vida media y mejorar la acción de este péptido. No obstante, dos estudios aleatorizados comparando la seguridad y eficacia de sitagliptina frente a placebo no demostraron diferencias en eficacia (mejoría de la esteatosis, NAS score o fibrosis) frente a placebo, en un estudio incluyendo 12 pacientes y otro con 50 pacientes prediabéticos o diabéticos con EHGNA60,61. Por tanto, no se recomienda el uso de inhibidores de la DPP-4 en el tratamiento de la esteatohepatitis no alcohólica.
AramcholAramchol es un ácido mixto (ácido graso-ácido biliar) sintético que se genera de la conjugación del ácido cólico (ácido biliar) y del ácido araquídico (ácido graso saturado), cuya función es inhibir la acción de la enzima stearoyl coenzyme A desaturase1 (SCD1) que es la encargada de catalizar la producción de grasas monoinsaturadas a partir de los ácidos grasos del hígado y de otros tejidos21. Esto produce una disminución de la reserva hepática de triglicéridos y de ésteres de ácidos grasos, que ha llevado a mostrar una reducción de esteatosis y mejoría en la resistencia a la insulina en modelos animales62. En sujetos obesos con esteatohepatitis se ha detectado una mayor actividad de enzima SCD1 frente a obesos con hígado sano. El aramchol ha sido evaluado en un ensayo clínico doble ciego7, controlado por placebo, de 60 pacientes con diagnóstico histológico de EHGNA (aunque solo 6 con esteatohepatitis) que fueron aleatorizados en grupos de 20 pacientes que recibieron 100mg o 300mg de aramchol, o placebo. Aramchol a dosis de 300mg fue seguro y efectivo en la reducción de la concentración hepática de grasa medida mediante espectroscopia por resonancia magnética tras 12 semanas de tratamiento. Además, el tratamiento con aramchol se asoció a un incremento en los niveles de adiponectina. Estudios en faseIII que demuestren el efecto del tratamiento sobre la lesión hepática valorada por biopsia definirán su lugar en la terapéutica en un futuro próximo.
Lugar de acción: estrés oxidativo e inflamaciónCenicriviroc es un antagonista dual de los receptores CCR2 y CCR5, los cuales están implicados en mecanismos de inflamación y fibrosis en la esteatohepatitis. CCR2/CCR5 participan en la migración e infiltración de monocitos proinflamatorios en el hígado, promoviendo daño hepático a través de la activación de las células de Kupffer63. Estudios en animales han demostrado la eficacia del fármaco. Cenicriviroc ha sido evaluado en el ensayo clínico CENTAUR (fármaco vs. placebo durante un año), que pretendía demostrar el efecto del fármaco en 289 pacientes en la mejoría NAS score >2 con al menos mejoría de balonización o inflamación lobulillar, sin empeoramiento de fibrosis. Los resultados preliminares del ensayo CENTAUR no mostraron diferencias en el objetivo principal (19% vs. 16%), aunque sí hubo mayor mejoría de la fibrosis hepática (20% vs. 10%), especialmente entre aquellos pacientes con NAS score >5 (24% vs. 10%). Además, cenicriviroc fue capaz de reducir los niveles de marcadores de inflamación como IL-1β, IL-6 y PCR11.
Las caspasas son enzimas que juegan un papel central en la apoptosis y la inflamación, teniendo un rol esencial en la patogénesis de la EHGNA. Emricasan es un potente inhibidor irreversible de las caspasas. En un ensayo clínico faseI, se demostró que emricasan era capaz de mejorar los biomarcadores de inflamación en pacientes con daño hepático64. En un ensayo clínico faseII, los niveles de ALT fueron significativamente más disminuidos en pacientes tratados que en pacientes que recibieron placebo12. Actualmente, el fármaco está en una siguiente fase para evaluar la mejoría de la histología hepática.
Apoptosis signal-regulating kinase1 (ASK1) es una proteína que interviene en la apoptosis bajo condiciones de estrés oxidativo65. Selonsertib es un inhibidor de la ASK1 que ha sido desarrollado para el tratamiento de la esteatohepatitis. En ensayo clínico faseII, el grupo de pacientes tratados con selonsertib se asoció a una reducción de al menos un grado de fibrosis hepática en un 43% frente a un 30% en el grupo placebo13. En la actualidad, hay un ensayo clínico en marcha para evaluar la eficacia de selonsertib 6mg vs. 18mg, con o sin simtuzumab.
Lugar de acción: barrera intestinalOrlistat es un inhibidor de la lipasa gastrointestinal que favorece la reducción de peso, disminuye el flujo de ácidos grasos libres al hígado y mejora la sensibilidad de la insulina sin producir efectos adversos hepatotóxicos. En un ensayo clínico doble ciego, aleatorizado y controlado con placebo en el que se evaluó su eficacia en la EHGNA, un total de 52 pacientes fueron aleatorizados a recibir el fármaco (120mg 3 veces al día) vs. placebo durante 6 meses. Se observó que los pacientes que habían tomado el fármaco mostraban una reducción más pronunciada de las cifras de ALT e insulina y una mayor reversibilidad de la esteatosis hepática examinada por ecografía, más allá de su efecto sobre la reducción del peso66. En una serie de 10 casos, Harrison et al.14 demostraron mejoría histológica en pacientes con pérdida de peso, la esteatosis mejoró en 6/10 y la fibrosis en 3/10 tras 6 meses de tratamiento. Por tanto, el tratamiento con orlistat, junto con el consejo dietético, induce pérdida de peso y mejoría hepática y metabólica. La administración de este fármaco se ha asociado con efectos adversos principalmente de tipo gastrointestinal, tales como esteatorrea, urgencia defecatoria, diarrea, flatulencia, dolor abdominal o fisuras anales; también se ha publicado algún caso de colelitiasis, pancreatitis aguda y en muy pocos casos hepatitis agudas de tipo colestásico y fallo hepático subagudo. A su vez se han descrito comorbilidades tales como hipertensión arterial, cetoacidosis diabética y alteraciones en el sistema nervioso y renal. Por estas últimas cuestiones (perfil de seguridad y la ausencia de valor añadido más allá de la pérdida de peso), su uso se ha limitado en el contexto de la EHGNA67.
Lugar de acción: fibrosis hepáticaLa fibrosis hepática es uno de los principales objetivos del tratamiento de EHGNA, tanto para evitar su aparición como para ser capaces de revertirla llegado el caso. Pocos son los fármacos que están siendo estudiados actualmente. El más destacable es el simtuzumab, que es un anticuerpo monoclonal contra la lysyl oxidase like-2 (LOXL2), una enzima profibrótica (activa TGF-β) abundante en los hepatocitos. Estudios in vitro han demostrado la eficacia de simtuzumab en la reducción y regresión de la fibrosis hepática68. Este fármaco ya ha mostrado resultados interesantes en la colangitis esclerosante primaria69. Actualmente, hay ensayos clínicos en marcha para evaluar la seguridad y eficacia de simtuzumab en pacientes con y sin cirrosis con EHGNA.
Otros mecanismos de acciónÁcidos grasos omega-3La esteatohepatitis no alcohólica está asociada con una disminución de ácidos grasos poliinsaturados70,71, los cuales se derivan de ácidos grasos esenciales y constituyen moléculas con alta actividad biológica. La administración de ácidos grasos omega-3 produce una clara mejoría en los niveles de triglicéridos en sangre y optimiza la sensibilidad periférica a la insulina, a la vez que reduce los efectos proinflamatorios a través de diferentes mecanismos de acción72-74. En un ensayo clínico aleatorizado, controlado con placebo y doble ciego, se analizó el papel del ácido etil-eicosapentaenoico en el tratamiento de la esteatohepatitis no alcohólica75. Se incluyeron 243 pacientes aleatorizados en 3 grupos, uno placebo y otros 2 a los que se les administraba diferentes dosis del fármaco (1.800 y 2.700mg/dl, respectivamente); de los cuales en 171 fueron finalmente analizados sus resultados. No se encontraron diferencias significativas entre los 3 grupos en cuanto al objetivo primario del estudio (disminución del NAS score sin empeoramiento de la fibrosis) ni tampoco en el análisis por subgrupos basado en la presencia o ausencia de diabetes. De la misma manera no se consiguió establecer diferencias en los objetivos secundarios del estudio (mejoría de la esteatosis, inflamación lobulillar, balonización de los hepatocitos y fibrosis). Recientemente, se ha publicado un metaanálisis considerando ensayos controlados por placebo y aleatorizados que evaluaban el uso de ácidos grasos poliinsaturados omega-3 en EHGNA, teniendo como objetivo el cambio en las cifras de ALT, AST, GGT, colesterol y triglicéridos. Por ello, los autores concluyeron que constituye un tratamiento efectivo en estos pacientes disminuyendo las cifras de ALT, colesterol y especialmente disminuyendo los triglicéridos, con aumento del colesterol HDL. Sin embargo, no se apreció mejoría de la fibrosis hepática evaluada mediante los niveles séricos de colágeno tipoIV76. Por todo ello, se necesitarían más estudios que avalaran la eficacia de esta terapia para frenar la evolución de la EHGNA.
OtrosSe están investigando nuevas dianas terapéuticas, con mecanismos de acción alternativos, como pueden ser los análogos de amilinas (pramlintida, davalintida), análogos de leptina (metreleptina), agonistas GLP-1 (exenatida, TTP-054), agonistas MC4R (RM-493), análogos de la oxintomodulina, antagonistas del neuropéptidoY (velneperit), bloqueantes de los endocannabinoides tipo1 (AM-6545), inhibidores de MetAP2 (beloranib), inhibidores la actividad lipasa (cetilistat) y vacunas frente a la obesidad (grelina, somatostatina y Ad36), que permitirán un tratamiento más personalizado de la obesidad77 (fig. 5).
Terapia quirúrgica y endoscópica
La terapia endoscópica de la esteatohepatitis no alcohólica está reservada a pacientes que no consiguen perder peso mediante intervención en el estilo de vida, ni tras el uso de fármacos que promueven la pérdida de peso como liraglutida. En pacientes con obesidad mórbida los porcentajes de fibrosis hepática avanzada (15%) y esteatohepatitis (25%) son elevados78. La cirugía bariátrica ha demostrado efectos beneficiosos sobre la esteatohepatitis no alcohólica y la fibrosis y además sobre otros factores de riesgo como la diabetes mellitus79, resistencia a la insulina80 o la hipertensión arterial81. Incluso puede incrementar la secreción de GLP-182 (asociado a la sensibilidad a la insulina), por lo que su efecto beneficioso iría más allá de la pérdida de peso o la mejoría del síndrome metabólico. Tanto la esteatosis como la esteatohepatitis no alcohólica revierten tras la cirugía bariátrica83, de manera coste-efectiva84. Estudios con biopsias pareadas han demostrado mejoría histológica en relación con la balonización, inflamación lobulillar y fibrosis, al año de la intervención. En concreto, la esteatosis y la inflamación portal pueden resolverse hasta en un 75% en 3 años tras la cirugía, mientras que la inflamación portal puede alcanzar el 50% y la esteatohepatitis el 90%. Aquellos pacientes con fibrosis grado2 (59%) y grado3 (29%) también pueden mejorar de forma significativa85. Bower et al. realizaron una revisión sistemática de 29 estudios que incluían pacientes obesos intervenidos mediante cirugía bariátrica. Los autores demostraron cómo la presencia de esteatosis, inflamación, balonización y fibrosis disminuía tras la intervención, así como evidenciaron una mejoría significativa en valores de ALT, AST y GGT86. No obstante, aunque los estudios publicados indican que la cirugía bariátrica se asocia a una mejoría significativa en pacientes con esteatohepatitis, el pequeño tamaño de los estudios y la heterogeneidad de la población incluida hace necesario la realización de ensayos clínicos con adecuado tamaño muestral. Una cuestión interesante es determinar si el efecto beneficioso de la cirugía bariátrica depende del tipo de intervención. La mayoría de los estudios publicados se basan en la cirugía gástrica en Y de Roux. En el estudio comparativo más importante hasta la fecha (incluyendo biopsias pareadas), Mathurin et al. incluyeron 381 pacientes intervenidos con técnicas restrictivas (banding gástrico) y malabsortivas (cirugía en Y de Roux, bypass biliointestinal) con biopsias repetidas al año y a los 5 años del procedimiento. Se objetivó mejoría en los parámetros histológicos de forma similar a estudios previos (esteatohepatitis del 27 al 14%), observando que dicha mejoría se producía especialmente durante el primer año. En el análisis comparativo entre técnicas no se observaron diferencias entre los distintos procedimientos87. Esta ausencia de diferencias ha sido visto en otros estudios88,89. Por tanto, no disponemos por el momento de datos robustos para recomendar un tipo de cirugía bariátrica sobre otro en pacientes con EHGNA, debiendo elegirse en función de su complejidad, balance riesgo-beneficio y extensión de la exclusión gastrointestinal.
Por su parte, la terapia endoscópica de la obesidad ha sido muy poco evaluada en pacientes con EHGNA. Uno de los escasos estudios publicados fue realizado por Lee et al., evaluando el papel del balón intragástrico (más dieta y ejercicio) frente a dieta y ejercicio en 18 pacientes con obesidad durante 6 meses. Los pacientes que se colocaron el balón intragástrico experimentaron mayor pérdida de peso y una mayor reducción del NAS score90. A pesar de estos datos alentadores, es necesario evaluar este tipo de intervenciones en cohortes mayores y con mayor seguimiento.
ConclusionesEl tratamiento de la esteatohepatitis no alcohólica abarca un amplio espectro de opciones terapéuticas desde medidas encaminadas a modificar el estilo de vida, la actividad física, la dieta y el deporte hasta el diseño de nuevas moléculas capaces de alterar los mecanismos patogénicos de la enfermedad. Los agonistas PPAR, FXR y GLP-1R conforman la base del futuro tratamiento de la esteatohepatitis no alcohólica. Moléculas dirigidas frente a la apoptosis, antioxidantes, o dirigidas frente a la respuesta inmune y la fibrosis deben buscar su espacio en la terapéutica de esta enfermedad en los próximos años. Por último, estos fármacos en desarrollo tienen dianas diferentes y algunas de ellas son complementarias, por lo que en un futuro los tratamientos combinados podrían mejorar los resultados comunicados hasta la fecha.
Conflicto de interesesNinguno.

