




Los fármacos inmunosupresores se encuentran entre los agentes farmacológicos con mayor potencial teórico de causar reacciones adversas, aunque la inducción de toxicidad hepática es una paradoja desde el punto de vista patogénico, toda vez que la respuesta del sistema inmunitario innato y adquirido se considera un episodio clave en la secuencia de acontecimientos que dan como resultado una lesión hepática de naturaleza química. La toxicidad hepática de los fármacos inmunosupresores es difícil de evaluar, ya que a veces se usan para tratar enfermedades hepáticas o junto con otros medicamentos que también pueden producir toxicidad hepática o se emplean en el contexto del trasplante hepático, en el que el rechazo o las complicaciones biliares pueden actuar como factores de confusión. Por otra parte, el tratamiento inmunosupresor puede favorecer la aparición de infecciones, que por sí mismas pueden causar daño hepático, o bien reactivar una hepatitis crónica viral latente. Los corticoides y los agentes inhibidores de la calcineurina excepcionalmente causan toxicidad hepática. El metotrexato a dosis elevadas y en pacientes con factores de riesgo puede inducir fibrosis avanzada y cirrosis. Los agentes tiopurínicos pueden ocasionar un espectro de lesiones hepáticas, incluyendo lesión hepatocelular o colestásica y alteraciones vasculares hepáticas. La leflunomida exhibe un elevado potencial hepatotóxico, especialmente si se combina con metotrexato. Los agentes antifactor de necrosis tumoral α se han asociado a toxicidad hepática en raras ocasiones, a menudo con autoanticuerpos detectables, y la mayoría de las reacciones, algunas de ellas graves, se han vinculado con infliximab, especialmente cuando se usa en pacientes con enfermedades reumatológicas.
Immunosuppressants are among the pharmacological agents with the greatest potential to cause adverse reactions, although induction of hepatotoxicity is paradoxical from the pathogenic point of view, since the response of the innate and acquired immune system is a key element in the chain of events leading to chemical liver damage.
Hepatotoxicity induced by immunosuppressants is difficult to evaluate since these drugs are sometimes used to treat liver diseases, or in combination with other drugs that can also cause hepatotoxicity, or in the context of liver transplantation, in which rejection or biliary complications can act as confounding factors. In addition, immunosuppressant therapy can favor the development of infections, which by themselves can cause liver damage, or reactivate latent chronic viral hepatitis. Corticosteroids and calcineurin inhibitors only exceptionally cause hepatotoxicity.
Methotrexate at high doses and in patients with risk factors can induce advanced fibrosis and cirrhosis. Thiopurine agents can cause a spectrum of hepatic lesions, including hepatocellular of cholestatic lesions, and hepatic vascular alterations. Leflunomide has high hepatotoxic potential, especially when combined with methotrexate. Anti-tumor necrosis factor-alpha agents have rarely been associated with hepatotoxicity, often with detectable autoantibodies, and most of the reactions – some severe – have been linked to infliximab, especially when used in patients with rheumatological diseases.
La toxicidad hepática se encuentra entre las más temidas reacciones adversas a medicamentos, tanto por su impacto en términos de morbimortalidad como por sus repercusiones económicas durante el proceso de desarrollo de los fármacos. Es, además, un problema desconcertante, debido al amplio número de sustancias capaces de inducir estas reacciones1, a la heterogeneidad de la presentación clínica2 y a la imposibilidad en el momento actual de establecer con certeza el diagnóstico en la inmensa mayoría de los casos. Hoy en día, el diagnóstico de hepatotoxicidad continúa siendo un reto en la práctica clínica y es habitualmente de exclusión. En general, el único modo de confirmar que un problema hepático es de naturaleza hepatotóxica es demostrar un recrudecimiento del daño hepático tras la reexposición al agente sospechoso3. La reexposición puede ser peligrosa, y sólo debería tenerse en cuenta si el fármaco se considera insustituible y tras obtener el consentimiento informado del paciente.
Los inmunosupresores se encuentran entre los grupos farmacológicos con mayor potencial teórico de inducir reacciones adversas, entre ellas las hepáticas, debido a su mecanismo de acción, y la comercialización en los últimos años de nuevos agentes, incluyendo los tratamientos biológicos, ha despertado preocupación al respecto.
Los inmunosupresores son fármacos capaces de suprimir la respuesta inmunológica a un estímulo antigénico, ya sea producido por un antígeno externo o interno. Los fármacos inmunosupresores se utilizan en la prevención del rechazo de los trasplantes y en una amplia serie de enfermedades autoinmunitarias, como la psoriasis, la enfermedad inflamatoria intestinal, la artritis reumatoide, la esclerosis múltiple y otras muchas enfermedades dermatológicas y sistémicas. En los últimos años se ha introducido una amplia variedad de agentes inmunosupresores para el tratamiento de diversas enfermedades autoinmunitarias (tabla 1). De acuerdo con su mecanismo de acción, los inmunosupresores se clasifican como:
- •
Fármacos que inhiben la activación de las células T y su respuesta a un órgano trasplantado o a estímulos antigénicos (ciclosporina, tacrolimus y sirolimus).
- •
Fármacos que inhiben la síntesis del ADN y el ARN y, por tanto, la división celular (azatioprina, micofenolato, ciclosporina y metotrexato).
- •
Corticoides que suprimen la inflamación asociada a la reacción inmunológica.
- •
Anticuerpos monoclonales que bloquean algunos factores implicados en el mecanismo de la reacción inmunológica, como las interleucinas o el factor de necrosis tumoral α (TNF-α). Entre ellos se encuentran comercializados el adalimumab, el infliximab, el basiliximab y otros productos de última generación obtenidos por recombinación genética (etanercept y alefacept).
- •
Fármacos que actúan sobre el factor estimulante de colonias (filgastrim, pegfilgastrim, molgramostin, lenogastrim, etc.).
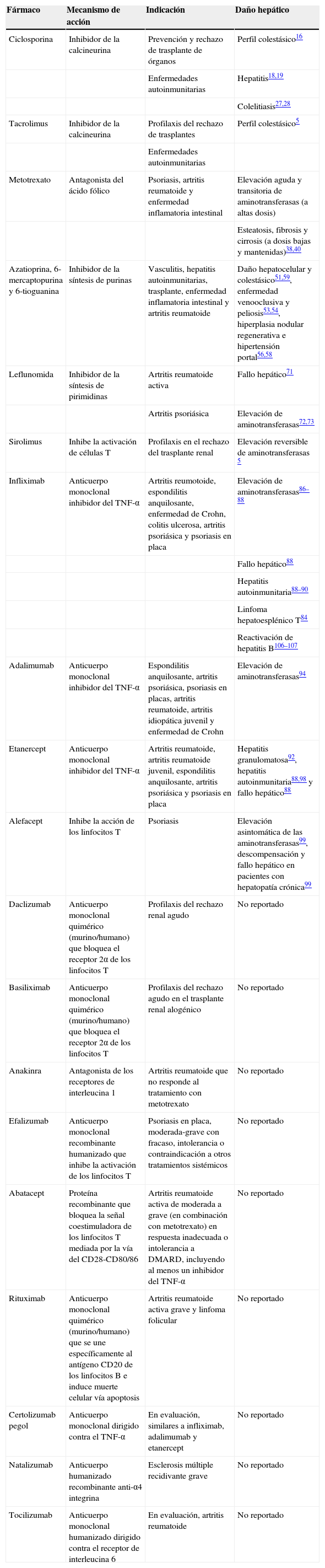
Indicaciones, mecanismo de acción y potencial hepatotóxico de los fármacos inmunosupresores
| Fármaco | Mecanismo de acción | Indicación | Daño hepático |
| Ciclosporina | Inhibidor de la calcineurina | Prevención y rechazo de trasplante de órganos | Perfil colestásico16 |
| Enfermedades autoinmunitarias | Hepatitis18,19 | ||
| Colelitiasis27,28 | |||
| Tacrolimus | Inhibidor de la calcineurina | Profilaxis del rechazo de trasplantes | Perfil colestásico5 |
| Enfermedades autoinmunitarias | |||
| Metotrexato | Antagonista del ácido fólico | Psoriasis, artritis reumatoide y enfermedad inflamatoria intestinal | Elevación aguda y transitoria de aminotransferasas (a altas dosis) |
| Esteatosis, fibrosis y cirrosis (a dosis bajas y mantenidas)38,40 | |||
| Azatioprina, 6-mercaptopurina y 6-tioguanina | Inhibidor de la síntesis de purinas | Vasculitis, hepatitis autoinmunitarias, trasplante, enfermedad inflamatoria intestinal y artritis reumatoide | Daño hepatocelular y colestásico51,59, enfermedad venooclusiva y peliosis53,54, hiperplasia nodular regenerativa e hipertensión portal56,58 |
| Leflunomida | Inhibidor de la síntesis de pirimidinas | Artritis reumatoide activa | Fallo hepático71 |
| Artritis psoriásica | Elevación de aminotransferasas72,73 | ||
| Sirolimus | Inhibe la activación de células T | Profilaxis en el rechazo del trasplante renal | Elevación reversible de aminotransferasas 5 |
| Infliximab | Anticuerpo monoclonal inhibidor del TNF-α | Artritis reumotoide, espondilitis anquilosante, enfermedad de Crohn, colitis ulcerosa, artritis psoriásica y psoriasis en placa | Elevación de aminotransferasas86–88 |
| Fallo hepático88 | |||
| Hepatitis autoinmunitaria88–90 | |||
| Linfoma hepatoesplénico T84 | |||
| Reactivación de hepatitis B106–107 | |||
| Adalimumab | Anticuerpo monoclonal inhibidor del TNF-α | Espondilitis anquilosante, artritis psoriásica, psoriasis en placas, artritis reumatoide, artritis idiopática juvenil y enfermedad de Crohn | Elevación de aminotransferasas94 |
| Etanercept | Anticuerpo monoclonal inhibidor del TNF-α | Artritis reumatoide, artritis reumatoide juvenil, espondilitis anquilosante, artritis psoriásica y psoriasis en placa | Hepatitis granulomatosa92, hepatitis autoinmunitaria88,98 y fallo hepático88 |
| Alefacept | Inhibe la acción de los linfocitos T | Psoriasis | Elevación asintomática de las aminotransferasas99, descompensación y fallo hepático en pacientes con hepatopatía crónica99 |
| Daclizumab | Anticuerpo monoclonal quimérico (murino/humano) que bloquea el receptor 2α de los linfocitos T | Profilaxis del rechazo renal agudo | No reportado |
| Basiliximab | Anticuerpo monoclonal quimérico (murino/humano) que bloquea el receptor 2α de los linfocitos T | Profilaxis del rechazo agudo en el trasplante renal alogénico | No reportado |
| Anakinra | Antagonista de los receptores de interleucina 1 | Artritis reumatoide que no responde al tratamiento con metotrexato | No reportado |
| Efalizumab | Anticuerpo monoclonal recombinante humanizado que inhibe la activación de los linfocitos T | Psoriasis en placa, moderada-grave con fracaso, intolerancia o contraindicación a otros tratamientos sistémicos | No reportado |
| Abatacept | Proteína recombinante que bloquea la señal coestimuladora de los linfocitos T mediada por la vía del CD28-CD80/86 | Artritis reumatoide activa de moderada a grave (en combinación con metotrexato) en respuesta inadecuada o intolerancia a DMARD, incluyendo al menos un inhibidor del TNF-α | No reportado |
| Rituximab | Anticuerpo monoclonal quimérico (murino/humano) que se une específicamente al antígeno CD20 de los linfocitos B e induce muerte celular vía apoptosis | Artritis reumatoide activa grave y linfoma folicular | No reportado |
| Certolizumab pegol | Anticuerpo monoclonal dirigido contra el TNF-α | En evaluación, similares a infliximab, adalimumab y etanercept | No reportado |
| Natalizumab | Anticuerpo humanizado recombinante anti-α4 integrina | Esclerosis múltiple recidivante grave | No reportado |
| Tocilizumab | Anticuerpo monoclonal humanizado dirigido contra el receptor de interleucina 6 | En evaluación, artritis reumatoide | No reportado |
DMARD: medicamentos antirreumáticos modificadores de la enfermedad; TNF-α: factor de necrosis tumoral α.
Muchos de estos fármacos causan frecuentes y a veces graves reacciones adversas, incluyendo el daño hepático tóxico. La capacidad de causar daño hepático de los inmunosupresores es una paradoja desde el punto de vista patogénico, toda vez que en muchos supuestos la respuesta del sistema inmunitario innato y adquirido se considera un evento clave en la secuencia de acontecimientos que dan como resultado toxicidad hepática de naturaleza química4. Este daño es, en todo caso, difícil de evaluar en el contexto de la inmunosupresión debido a que los inmunosupresores a menudo se emplean junto con otros medicamentos que también pueden producir toxicidad hepática o incluso se utilizan para tratar enfermedades hepáticas o en el contexto del trasplante hepático, en el que el rechazo o las complicaciones biliares pueden actuar como factores de confusión5. Por otra parte, el tratamiento inmunosupresor puede favorecer la aparición de infecciones que por sí mismas pueden causar daño hepático o reactivar una hepatitis crónica viral latente. En la presente revisión se abordará la toxicidad hepática inducida por los fármacos inmunosupresores con especial atención a las nuevas moléculas.
CorticoidesLos corticoides fueron los primeros agentes inmunosupresores introducidos en clínica. Constituyen un pilar básico del tratamiento de una gran variedad de enfermedades autoinmunitarias y desempeñan un papel fundamental en los regímenes inmunosupresores que siguen al trasplante de órganos sólidos y en el tratamiento del rechazo. La complicación aguda más grave derivada de la retirada brusca de los esteroides tras una administración prolongada es la insuficiencia suprarrenal aguda6. Otros efectos adversos incluyen hipertensión arterial, hipercolesterolemia, intolerancia a la glucosa, insomnio, labilidad emocional, cuadros psicóticos, cataratas, osteopenia con aplastamientos vertebrales, diabetes y cambios estéticos, como “cara de luna llena”, “giba de búfalo”, acné e hirsutismo6. Estos efectos adversos aparecen en el 80% de los pacientes tras 2 años de tratamiento y desaparecen al retirar los corticoides.
En lo que se refiere a los efectos sobre el hígado, altas dosis de glucocorticoides pueden producir esteatosis hepática al favorecer la movilización y la redistribución grasa, incrementando en el plasma los ácidos grasos libres inhibiendo la esterificación de ácidos grasos en el hígado7,8. Todo ello, junto con el incremento de la resistencia insulínica, favorece la esteatosis macrovesicular9. Dosis más bajas de glucocorticoides (10–15mg/día) cuando se administran durante períodos prolongados pueden causar también esteatosis5.
Los glucocorticoides pueden producir también esteatohepatitis no alcohólica en individuos predispuestos por aumento de la resistencia insulínica10 y al favorecer la obesidad central pueden producir hipertrigliceridemia y diabetes11. Por otro lado, la lesión hepática aguda de carácter idiosincrásico debida a corticoides es un evento inusual y, en general, los parámetros bioquímicos de función hepática permanecen dentro de los rangos normales. Sin embargo, se han descrito algunos casos aislados de hepatotoxicidad tras la administración de metilprednisolona11, en uno de estos pacientes (una paciente con esclerosis múltiple) se documentó que la lesión hepatocelular reaparecía pocos días tras la administración de pulsos de metilprednisolona intravenosa12, lo que refuerza la atribución de causalidad.
CiclosporinaLa ciclosporina es un polipéptido cíclico formado por 11 aminoácidos producido a partir del hongo Beauveria nlyea. Es un agente inhibidor de la calcineurina, lipofílico, con fuertes propiedades hidrofóbicas, cuya absorción es variable. La ciclosporina actúa inhibiendo una gran cantidad de citocinas proinflamatorias, especialmente la interleucina (IL) 2 y el IFN-γ. También actúa sobre la inmunidad celular; así, se ha observado un efecto antiproliferativo de los linfocitos, más marcado en los T-helper CD4+ que en los T-supresores CD8+13. No interfiere con la actividad de los monocitos, los granulocitos o los macrófagos.
La ciclosporina ha tenido un amplio uso clínico desde su introducción en la década de 1970, especialmente en la prevención del rechazo en el trasplante de órganos, y más recientemente se ha empleado en el tratamiento de diversas enfermedades autoinmunitarias5. Los más frecuentes efectos adversos derivados de este fármaco son la nefrotoxicidad, la hipertensión arterial y la neurotoxicidad. Menos frecuentes son la toxicidad hepática, las convulsiones (favorecidas por la hipomagnesemia y la hipocolesterolemia), las alteraciones hematológicas, la anafilaxia y las infecciones oportunistas (neumonía por Pneumocystis carinii y esofagitis herpética)14,15.
En la práctica clínica, el daño hepático causado por la ciclosporina se caracteriza por un aumento sérico de los niveles de fosfatasa alcalina y una leve elevación de las aminotransferasas y de la bilirrubina conjugada. Estas alteraciones bioquímicas aparecen generalmente entre la segunda semana y el tercer mes tras el inicio del tratamiento, y tienden a normalizarse tras reducir la dosis del fármaco16. Histológicamente, la afectación hepática secundaria a ciclosporina se caracteriza por una hipertrofia del epitelio ductal biliar, vacuolas citoplásmicas y la presencia de un material “espumoso” en el interior de los sinusoides hepáticos17.
Se han descrito casos graves de hepatitis colestásica inducida por ciclosporina, aunque se trata de un efecto adverso poco frecuente18,19. Son factores de riesgo para su aparición la existencia previa de ictericia o colestasis, la infección por el virus de la hepatitis C (VHC) y la nutrición parenteral20,21. La prevención se basa en la monitorización de los niveles séricos de ciclosporina y el control de la función renal, fundamentalmente en pacientes con daño hepático subyacente o con nutrición parenteral22–24. Por otro lado, la ciclosporina es metabolizada por el citocromo P-450, y los fármacos que inhiben esta vía metabólica pueden incrementar los niveles séricos de la ciclosporina y aumentar con ello el riesgo de toxicidad hepática25.
Se ha indicado que la administración de ácido ursodesoxicólico podría ser beneficiosa para prevenir o acelerar la resolución de la lesión hepática inducida por ciclosporina, pero serían necesarios ensayos clínicos controlados para establecer con certeza su papel en este contexto26.
Finalmente, el tratamiento con ciclosporina se ha asociado al desarrollo de colelitiasis, fundamentalmente en pacientes con más de 2 años de tratamiento27,28. El riesgo de formación de cálculos biliares está aumentado en el páncreas diabético y en pacientes con trasplante renal, en los que la prevalencia puede alcanzar hasta el 30%29.
TacrolimusEl tacrolimus, un inhibidor calcineurínico que bloquea la activación de los linfocitos-T, empezó a usarse como profilaxis del rechazo de trasplantes y actualmente también se utiliza en el tratamiento de enfermedades autoinmunitarias.
La hepatotoxicidad por tacrolimus es relativamente poco frecuente. Se han descrito casos de colestasis que han revertido al cambiar tacrolimus por ciclosporina. También se han descrito casos en los que la colestasis asociada al tratamiento con ciclosporina se ha resuelto al cambiar a tacrolimus5. Recientemente se ha descrito un caso de colestasis asociada a tacrolimus que no respondió al cambio a ciclosporina, y se resolvió sólo cuando descendieron los niveles en sangre del fármaco30.
SirolimusEs un antibiótico macrocíclico con efectos inmunosupresores, antiproliferativos, antifúngicos, antifibróticos y antineoplásicos. A pesar de tener efectos secundarios graves, como mielosupresión, úlceras aftosas y neumonitis, la hepatotoxicidad por sirolimus es poco frecuente y únicamente se han descrito elevaciones reversibles de aminotransferasas. En algunos estudios también se ha asociado con un incremento del riesgo de trombosis de la arteria hepática tras trasplante hepático5,31.
MetotrexatoEl metotrexato es un antagonista del ácido fólico, usado inicialmente como agente quimioterápico en las neoplasias hematológicas (principalmente leucemias). Posteriormente empezó a utilizarse con buenos resultados por su efecto inmunosupresor en el tratamiento de la psoriasis, la artritis reumatoide y la enfermedad inflamatoria intestinal32. El metotrexato es un análogo de los folatos. Compite con el ácido folínico y con el folato sérico para penetrar en la célula. Una vez en el interior, la enzima folato-poliglutamasa sintasa transforma al metotrexato en poliglutamatos, metabolitos de larga vida media, causantes de su actividad antifolato y de sus efectos citotóxicos33. Intracelularmente, el metotrexato y sus metabolitos inhiben diferentes enzimas dependientes de folatos (principalmente la dihidrofolato reductasa, enzima que interviene en la síntesis de purinas y pirimidinas, necesarias para la formación de ARN y ADN)34, así como la conversión de homocisteína a metionina. El exceso de homocisteína puede producir lesión hepática por varios mecanismos, que incluyen la sensibilización de la célula al efecto citotóxico de agentes o situaciones que inducen estrés oxidativo35. En un contexto de estrés oxidativo del retículo endoplásmico, la acumulación de proteínas erróneamente almacenadas debido a la disrupción de anillos disulfuro puede inducir la activación de factores de transcripción, entre ellos la proteína reguladora de esteroles, causante de la síntesis de lípidos para generación de las membranas del retículo endoplásmico, lo que resulta en esteatosis hepática36, y además de forma directa puede inducir apoptosis36. Finalmente, la homocisteína puede activar citocinas proinflamatorias, y el resultado de una amplia gama de insultos sobre la célula estrellada es la activación de ésta y la producción de colágeno.
La toxicidad del metotrexato es dependiente de la dosis. Es por esto que se ve influenciada por factores que afecten su absorción, distribución y excreción. A altas dosis produce elevaciones agudas y transitorias de la aspartato aminotransferasa (AST), así como mielosupresión, reacciones mucocutáneas, neumonitis y alteraciones gastrointestinales (anorexia, náuseas y diarrea). A dosis bajas y mantenidas en el tiempo, como las utilizadas en el tratamiento de la psoriasis, la artritis reumatoide y la enfermedad inflamatoria intestinal, produce alteraciones hepáticas que van desde anormalidades analíticas (elevación de AST, alanina aminotransferasa [ALT] y fosfatasa alcalina [FA]) hasta hepatopatía crónica, fibrosis y cirrosis. También se han descrito casos de fallo hepático fulminante. Roenigk et al37 realizaron en la década de 1970 una clasificación estandarizada del daño hepático asociado a metotrexato según los hallazgos histológicos que se sigue considerando útil. La toxicidad hepática depende de la dosis y de la frecuencia de la administración. Los estudios iniciales estimaban frecuencias elevadas (entre el 11 y el 26%) de cirrosis en pacientes con psoriasis que recibían largos tratamientos con el compuesto38, lo que justificaría la realización de biopsias hepáticas seriadas. Sin embargo, tras un mejor conocimiento de los factores de riesgo y una mayor optimización de la dosis, recientemente se ha reportado que la incidencia de esta reacción adversa hepática es inferior (entre el 0 y el 10%)39.
La relación entre el desarrollo y el grado de fibrosis con la dosis acumulada de metotrexato está bien establecida. La administración de dosis semanales superiores a 20mg o la administración —actualmente abandonada— diaria o cada 48 h se asocian a mayor incidencia de cirrosis (3–26%) que la administración de una dosis semanal máxima de 20mg (0–4%). En general, se aprecia un mayor riesgo de fibrosis hepática cuando la dosis acumulada es superior a 1,5g40, aunque en algunos estudios esta asociación no ha sido estadísticamente significativa41 y otros autores han puesto en cuestión este hecho42. Los principales factores de riesgo identificados para el desarrollo de esta complicación son el abuso de alcohol, la existencia de enfermedades hepáticas previas (como hepatitis B y hepatitis C), la edad, la obesidad y la diabetes43. Dado que el estrés del retículo endoplásmico puede ser un vínculo mecanístico entre obesidad, resistencia insulínica y diabetes de tipo 244,45 y que esta alteración podría subyacer en la lesión hepática por alcohol y en la interacción entre consumo de alcohol y hepatitis viral46, se ha especulado con la posibilidad de que el metotrexato al causar per se estrés del retículo endoplásmico favorecería particularmente la lesión hepática en el contexto de otras situaciones que, asimismo, indujesen este estado, como obesidad, diabetes, consumo de alcohol o hepatitis crónica viral47.
Recientemente, se ha puesto en cuestión la utilidad de la biopsia hepática en el manejo clínico (indicación y monitorización) de pacientes en tratamiento con metotrexato, dado que ninguno de una serie de 69 pacientes con psoriasis a los que se les realizaron biopsias durante el tratamiento desarrolló cirrosis en el período de 6 años analizado y en ningún caso los hallazgos histológicos obligaron a suspender el tratamiento48.
Azatioprina y otros agentes tiopurínicosLa azatioprina y sus análogos (6-mercaptopurina y 6-tioguanina) son inmunomoduladores tiopurínicos con potente efecto inmunosupresor y citostático, empleados en una amplia variedad de enfermedades autoinmunitarias, como la vasculitis, la hepatitis autoinmunitaria, el lupus eritematoso sistémico, la artritis reumatoide y la enfermedad inflamatoria intestinal, así como en el trasplante y en neoplasias como la leucemia linfoblástica.
La eficacia clínica de los inmunomoduladores tiopurínicos está limitada por la aparición de efectos adversos que obligan a la retirada del tratamiento en un 15–30% de los pacientes49,50. El acúmulo intracelular de 6-tioguanina, como consecuencia del fallo de la enzima tiopurina metiltransferasa (TPMT) que deriva la metabolización de la 6-mercaptopurina a otras vías enzimáticas, es el causante de los efectos citotóxicos de este fármaco. Los efectos adversos aparecen en el 10% de los pacientes que toman 50mg/día de azatioprina, y se pueden dividir en independientes de la dosis o idiosincráticos (rash, fiebre, diarrea y pancreatitis) y dependientes de la dosis o tóxicos (náuseas y mielotoxicidad). La hepatotoxicidad se ha descrito tanto como un efecto adverso alérgico como un efecto dependiente de la dosis5. Aunque el daño hepático manifiesto es raro, se ha descrito con más frecuencia en el sexo masculino y tras el trasplante renal51.
La hepatotoxicidad inducida por azatioprina puede manifestarse de forma variada, como lesión hepatocelular, colestásica y mixta, y como daño en el endotelio vascular que puede seguirse de un incremento de la presión portal, hiperplasia nodular regenerativa52, enfermedad venooclusiva y peliosis hepática53,54, que se suponen diferentes estadios del mismo daño. En pacientes con enfermedad inflamatoria intestinal la incidencia anual de toxicidad hepática inducida por azatioprina o 6-mercaptopurina, en una revisión sistemática reciente, se ha estimado que oscila entre un 1,4% en estudios retrospectivos y más de un 10% en estudios prospectivos55. El análogo 6-tioguanina en tratamientos prolongados en la leucemia linfoblástica y la enfermedad inflamatoria intestinal ha inducido frecuentemente enfermedad venooclusiva, hiperplasia nodular regenerativa e hipertensión portal56–58 y sus complicaciones con una relación temporal plausible58.
El cuadro clínico de la hepatotoxicidad inducida por los inmunosupresores tiopurínicos se ha descrito tradicionalmente como una hepatitis aguda colestásica, aunque el espectro de lesiones puede ser más amplio, con predominio de citolisis e incluso hipertransaminasemia asintomática. En una serie de 7 pacientes con trasplante renal con toxicidad hepática atribuida a azatioprina, 5 desarrollaron colestasis centrolobular asociada a esteatosis y 3 tuvieron dilatación sinusoidal. La imputabilidad de azatioprina se reforzó porque hubo 3 casos con reexposición positiva59.
Esta variabilidad de expresión clínica podría ser el reflejo de distintos mecanismos de acción involucrados en la toxicidad hepática. En realidad, cualquiera de los componentes de la azatioprina —6-mercaptopurina y el grupo imidazol y sus metabolitos (6-tioguanina)— podría desempeñar un papel importante en el desarrollo de la hepatotoxicidad. El hecho de que 6-mercaptopurina se haya vinculado con lesión hepatocelular, colestásica y endotelial sinusoidal y 6-tioguanina se haya vinculado con enfermedad venooclusiva da soporte a esta hipótesis60. El mecanismo de producción de lesión hepática por azatioprina y 6-mercaptopurina en el animal de experimentación es diferente según se empleen dosis terapéuticas o supraterapéuticas; a dosis altas de azatioprina en modelos de cultivo celular de roedores se ha apreciado una intensa depleción de glutatión, tanto en células endoteliales sinusoidales como en hepatocitos, y podía modularse la lesión disminuyendo o aumentando la concentración de glutatión del medio de cultivo61,62. Este mecanismo de lesión se ha atribuido a la conjugación del anillo nitroimidazol al glutatión merced a la acción de la glutatión-S-transferasa63. El mecanismo de producción de lesión hepática con dosis terapéuticas de azatioprina y 6-mercaptopurina podría ser la oncosis celular (necrosis celular por deenergización) inducida por el estrés oxidativo mediado por la xantina-oxidasa que es seguido de depleción de glutatión y ATP mitocondrial64. Este mecanismo no se relaciona con el anillo imidazol y se previene experimentalmente con la administración de alopurinol (un inhibidor de la xantina-oxidasa) y de trolox (un análogo soluble en agua de la vitamina E)54.
Las manifestaciones clínicas pueden ir desde casos asintomáticos hasta una amplia variedad de síntomas, como malestar general, artralgias, fatiga, fiebre, anorexia, dolor abdominal, náuseas, vómitos, diarrea, pérdida de peso y prurito. En casos graves se han descrito insuficiencia hepática con alteraciones de la coagulación y complicaciones derivadas de la hipertensión portal. Los hallazgos de laboratorio pueden mostrar incremento de las aminotransferasas y las enzimas de colestasis con o sin elevación de la bilirrubina60.
Una estrategia para reducir la toxicidad de los inmunosupresores tiopurínicos es detectar a los individuos que metabolizan deficientemente las tiopurinas determinando los genotipos de la TPMT. En pacientes con enfermedad de Crohn en tratamiento con azatioprina, una concentración eritrocitaria de metabolitos nucleótidos de 6-tioguanina (E-6-TGN) superior a 230pmol/8×10 eritrocitos (RBC) se asocia a respuesta al tratamiento, mientras que una concentración eritrocitaria de metabolitos metilados superior a 5.000pmol/8×10 RBC se ha correlacionado con hepatotoxicidad. La TPMT es causante de la formación de metabolitos metilados con niveles más bajos de E-6-TGN, y el genotipado de TPMT es muy útil para guiar la dosificación correcta de compuestos tiopurínicos65. Aunque el desarrollo de hepatotoxicidad parece relativamente independiente de los polimorfismos de TPMT, el análisis farmacogenético debería ser rutinario para prevenir las toxicidades hematológicas y ajustar mejor la dosis en pacientes que requieran tratamientos prolongados con dosis plenas66. La monitorización del perfil hepático es otra estrategia aconsejada para reducir la incidencia de toxicidad hepática en pacientes con enfermedad inflamatoria intestinal55; una elevación de escasa magnitud de las aminotransferasas puede permitir mantener el tratamiento porque las alteraciones son a menudo reversibles. Cuando las alteraciones son de mayor entidad, debería reducirse la dosis de azatioprina/6-mercaptopurina un 50% con estrechos controles clínicos y analíticos. Con esta estrategia los valores de aminotransferasas frecuentemente se normalizan y puede restaurarse la dosis inicial. Sin embargo, en pacientes que desarrollan ictericia debe suspenderse de inmediato el agente tiopurínico dado que la evolución puede ser grave y progresiva. Por último, la gravedad potencial de la hepatotoxicidad inducida por 6-tioguanina desaconseja su utilización fuera del contexto de ensayos clínicos55.
En el animal de experimentación, la administración de componentes protectores hepáticos, como la N-acetilcisteína y la aminoguanidina, parecen disminuir el riesgo si se toman previamente o durante el tratamiento67.
LeflunomidaLa leflunomida es un agente inmunomodulador isoxazólico que inhibe la dihidroorotato deshidrogenasa (una enzima que participa en la síntesis de novo de la pirimidina) e inhibe de forma reversible la proliferación de los linfocitos autoinmunitarios y activados68. Tras su administración, la leflunomida sufre metabolismo hepático y da lugar a un metabolito activo (A771726) que es el principal causante de la actividad farmacológica de ésta. La FDA (Food and Drug Administration) aprobó a la leflunomida en septiembre de 1999 y está indicada en el tratamiento de la artritis reumatoide activa (como medicamentos antirreumáticos modificadores de la enfermedad) y en la artritis psoriásica69. Los efectos adversos asociados a leflunomida se deben fundamentalmente a su prolongada vida media (entre 15 y 18 días). Diferentes ensayos clínicos demostraron que el tratamiento con leflunomida se asocia a un aumento de los niveles séricos de aminotransferasas en un número significativo de individuos tratados, pero, por lo general, se trata de incrementos leves (<2 X límite superior de la normalidad [LSN]) que se revierte aun manteniendo el tratamiento68,70. Sin embargo, en marzo del 2001 la Agencia Europea de Evaluación de Productos Medicinales (EMEA) advirtió sobre el potencial daño hepático grave en pacientes tratados con leflunomida. Con una exposición total estimada de 104.000 pacientes/año se han notificado 296 casos de reacciones hepáticas, 129 de éstas se consideraron graves, entre ellas 2 casos de cirrosis hepática y 15 de insuficiencia hepática (9 de ellas mortales). Las reacciones hepáticas aparecieron en los primeros 6 meses de tratamiento. En muchos casos estaban presentes factores de confusión. Así, entre los 129 casos graves, 101 (78%) estaban recibiendo simultáneamente otras medicaciones hepatotóxicas y 33 (27%) presentaban otros factores de riesgo, como abuso de alcohol, alteraciones de la función hepática, insuficiencia cardíaca aguda, neumopatías graves o carcinoma pancreático; entre los pacientes con pruebas funcionales hepáticas anormales, el 58% también estaba recibiendo metotrexato o antiinflamatorios no esteroideos71. La asociación con otros fármacos potencialmente hepatotóxicos, como metotrexato, incrementa considerablemente (hasta en un 50%) la incidencia de alteraciones del perfil hepático en pacientes en tratamiento con leflunomida72,73. En un reciente análisis de pacientes con artritis reumatoide y artritis psoriásica enrolados en el Consortium of Rheumatology Researchers of North America, las elevaciones de AST/ALT superiores a 2 X LSN ocurrieron en un 1–2% de pacientes tratados con leflunomida en monoterapia y en un 5% de los que recibían la combinación, y el riesgo aumentaba desde 2,91 veces en los pacientes que recibían una dosis de metotrexato de 10–17,5mg/semana a 3,98 veces en los pacientes que recibían una dosis superior o igual a 20mg/semana73.
El mecanismo de toxicidad hepática de leflunomida es desconocido, pero debido a su elevada frecuencia actualmente se recomienda que en los pacientes tratados con este antirreumático en monoterapia se realice una determinación de la ALT basal y a intervalos mensuales durante 6 meses, período tras el que, si estos niveles permanecen estables, se determinará cada 6–8 semanas. En los pacientes que desarrollan elevaciones leves (2–3 X LSN) se recomienda una reducción de dosis de leflunomida (10mg/día). Si los niveles de ALT continúan elevados (2–3 X LSN) a pesar de la reducción de la dosis, se recomienda una biopsia hepática en aquellos pacientes que continúan con el tratamiento. Finalmente, si los niveles de ALT superiores a 3 X LSN persisten a pesar de la reducción de la dosis se recomienda suspender el tratamiento con leflunomida e iniciar tratamiento de “lavado” con colestiramina5.
Por todo esto, la leflunomida está contraindicada en pacientes con alteración de la función hepática y no es aconsejable su uso concomitante con metotrexato u otros fármacos hepatotóxicos, pues se asocia a un aumento del riesgo de reacciones hepáticas graves.
Antagonistas del factor de necrosis tumoral: infliximab, adalimumab y etanerceptLos nuevos tratamientos biológicos han supuesto un importante avance en el tratamiento de numerosas enfermedades de base autoinmunitaria. Estos fármacos son moléculas que actúan por distintos mecanismos como inhibidores del TNF-α. Las actividades biológicas atribuidas al TNF-α que estas nuevas moléculas bloquearían incluyen la inducción de varias citocinas inflamatorias, como las IL 1 y 6, el incremento de la migración leucocitaria al aumentar la permeabilidad del endotelio, la expresión de moléculas de adhesión por leucocitos y células endoteliales, la activación de neutrófilos y eosinófilos, la proliferación de fibroblastos y la síntesis de prostaglandinas74. Los fármacos anti-TNF-α se utilizan para el tratamiento de enfermedades inflamatorias crónicas tanto reumatológicas como digestivas.
El infliximab es un anticuerpo monoclonal quimérico (humano/murínico) tipo IgG1 dirigido contra el TNF-α. El infliximab neutraliza la actividad biológica del TNF-α y se fija a las áreas de alta afinidad y transmembrana de éste, e impide que pueda unirse a sus receptores; además, neutraliza las células productoras de TNF-α e induce la apoptosis de los linfocitos T activados75. El infliximab se emplea actualmente en el tratamiento de la artritis reumatoide, la espondilitis anquilosante, la enfermedad de Crohn y la colitis ulcerosa, la artritis psoriásica y la psoriasis en placas76–78.
Los efectos adversos más frecuentes son cuadro seudogripal, infecciones secundarias a inmunodepresión, hipertensión, fatiga, artromialgias, alteraciones gastrointestinales inespecíficas y reacciones anafilácticas. Otras alteraciones menos frecuentes son arritmias cardíacas, insuficiencia cardíaca, anemia, leucocitopenia, trombocitopenia y aumento de la incidencia de determinados tipos de neoplasias79.
El adalimumab es un anticuerpo monoclonal recombinante tipo IgG1 que inhibe la acción del TNF-α al bloquear sus receptores de superficie celulares80.
Está indicado su uso en el tratamiento de la espondilitis anquilosante, la artritis psoriásica, la psoriasis en placas, la artritis reumatoide, la artritis idiopática juvenil y la enfermedad de Crohn81. Los efectos adversos más destacados son reacción local en la zona de inyección, síndrome seudogripal, hipertensión arterial, hiperlipidemia y aumento del riesgo de infecciones debido al efecto inmunosupresor79.
El etanercept es una proteína recombinante derivada del ADN compuesta por receptor del TNF-α unido a la fracción Fc de la inmunoglobulina G1 (IgG1). Actúa uniéndose al TNF-α e impide así su interacción con los receptores de superficie celulares82.
Su administración está indicada en la artritis reumatoide, la artritis reumatoide juvenil, la espondilitis anquilosante, la artritis psoriásica y la psoriasis en placas83. Sus efectos adversos más destacados son similares a los de adalimumab79. Otros inmunosupresores con su mecanismo de acción e indicaciones se recogen en la tabla 1.
Hepatotoxicidad por antagonista del factor de necrosis tumoral αUno de los principales efectos adversos potenciales del tratamiento con anti-TNF es el incremento del riesgo de linfomas y de algunos tumores sólidos. Recientemente se documentaron casos de linfoma hepatoesplénico de células T en pacientes jóvenes con enfermedad de Crohn en tratamiento con infliximab, los cuales habían recibido previamente tratamiento con tiopurinas. Se trata de una rara y en ocasiones mortal forma de linfoma no hodgkiniano que suele afectar a adultos y a niños jóvenes84.
La hepatotoxicidad, en cambio, es rara y la mayoría de los casos reportados se han asociado al tratamiento con infliximab85; aparece con mayor frecuencia en pacientes con enfermedades reumatológicas, especialmente en la artritis psoriásica. Durante los ensayos clínicos previos a su aprobación por la FDA la proporción de casos de elevación de aminotransferasas de magnitud leve a moderada (<3 X LSN) fue mayor en el grupo de infliximab (37%) que en el grupo de placebo, aunque en este último era también elevada (29%)86. De igual modo, en el ensayo ACCENT que evaluaba la eficacia a largo plazo de infliximab en pacientes con enfermedad de Crohn, se detectaron elevaciones de aminotransferasas en un 42% de los pacientes87. No se observaron incidencias de ictericia o fallo hepático durante el desarrollo clínico del fármaco. Sin embargo, con posterioridad a su comercialización se han reportado episodios de hepatotoxicidad clínicamente expresivos atribuidos a infliximab, incluyendo un total de 31 casos a la FDA moderadamente bien documentados, aunque con otras causas potenciales de daño hepático, como sepsis, infección viral o medicación hepatotóxica concomitante88. El período de latencia osciló entre 2 semanas y 12 meses, y se registraron casos de muerte y trasplante hepático88. Algunos de los casos documentados de hepatotoxicidad por infliximab tenían características de hepatitis autoinmunitaria, incluyendo un elevado título de anticuerpos antinucleares88–90; sin embargo, el tratamiento con antagonistas del TNF-α se ha asociado al desarrollo de estos anticuerpos incluso en ausencia de daño hepático, por lo que el significado de éstos no queda claro88. Sobre la base, pues, de estos datos poscomercialización, la FDA advirtió sobre el riesgo potencial de daño hepático asociado al tratamiento con infliximab88.
El tratamiento con etanercept también se ha vinculado con daño hepático; 19 casos de fallo hepático enviados a la FDA se atribuyeron a etanercept, aunque otras causas potenciales de lesión hepática estaban presentes en todos los pacientes88. Muy pocas publicaciones vinculan a etanercept con hepatotoxicidad; una paciente con artritis psoriásica desarrolló hepatitis al recibir etanercept, pero otros fármacos potencialmente hepatotóxicos y una posible sepsis eran factores de confusión evidentes91. Un caso de hepatitis granulomatosa documentado con biopsia imputable a etanercept se ha publicado recientemente92. El daño hepático relacionado con etanercept también se ha descrito con características autoinmunitarias88,93.
Finalmente, el potencial hepatotóxico de adalimumab parece bajo, con casos aislados publicados94. En una serie reciente de 47 pacientes con enfermedad de Crohn refractaria que recibieron dosis en escalada de adalimumab hubo un 8% de retiradas por efectos adversos, pero no se registró ningún caso de hepatotoxicidad95.
Algunos casos report indican la ausencia de hepatotoxicidad cruzada entre infliximab y etanercept96,97 y entre adalimumab y etanercept94, aunque la experiencia es anecdótica y, por tanto, no concluyente5. Sin embargo, la plausibilidad de este hecho se sostiene por el diferente mecanismo de acción de las moléculas (anticuerpos anti-TNF-α, en el caso de infliximab y adalimumab, y proteína de fusión del receptor del TNF-α, en el caso de etanercept) así como por la frecuente utilidad de este último en casos en los que han fallado los primeros98.
El alefacept fue el primer agente biológico aprobado por la FDA para el tratamiento de la psoriasis. Se trata de una proteína de fusión humana que se une al receptor CD2 inhibiendo la acción de los linfocitos T. Se han reportado a la FDA casos de elevación asintomática de aminotransferasas y de descompensación y fallo hepático en pacientes con hepatopatía crónica99.
Para otros nuevos agentes inmunosupresores no hay evidencia suficiente de potencial hepatotóxico (tabla 1).
Otro aspecto de preocupación con los nuevos tratamientos biológicos, como ocurre con inmunosupresores clásicos, es la posible reactivación de una hepatitis crónica viral. Los pacientes con infección por los virus de la hepatitis B y C exhiben elevados niveles en sangre de citocinas, incluyendo TNF-α100,101, y aunque el papel del TNF-α en la progresión de la enfermedad hepática no está claro, podría ser teóricamente beneficiosa su inhibición, ya que se ha indicado que el TNF-α incrementa la apoptosis hepatocitaria y finalmente la fibrosis en la hepatitis viral102,103. Sin embargo, la incapacidad de producir cantidades apropiadas de TNF-α o un desequilibrio en las células T facilitadoras circulantes se asocia a un aclaramiento viral disminuido104, constituyendo un riesgo potencial en pacientes con hepatitis viral crónica. Existe muy poca información acerca del efecto de los fármacos anti-TNF-α sobre la inflamación hepática o la replicación viral en pacientes con hepatitis crónica B o C subyacente. En un estudio retrospectivo reciente de 8 pacientes con hepatitis crónica C y 3 pacientes con hepatitis crónica B que recibían fármacos anti-TNF-α debido a una artritis reumatoide, únicamente en 2 casos se observaron incrementos transitorios de aminotransferasas durante el tratamiento y únicamente en un paciente (con infección por el virus C) se produjo un incremento de la viremia105. En todo caso, es la reactivación de la infección viral B la que es de especial trascendencia. Así, se ha reportado un paciente infectado por el virus de la hepatitis B, que desarrolló una hepatitis fulminante al recibir infliximab por enfermedad de Still del adulto refractaria106, y se ha reportado un paciente que era portador crónico del antígeno de superficie de la hepatitis B (AgHBs) no identificado y desarrolló una hepatitis subfulminante al recibir una infusión de infliximab por enfermedad de Crohn107.
Es imperativo, por tanto, efectuar un cribado para los virus de la hepatitis en pacientes que vayan a recibir un tratamiento inmunosupresor con anti-TNF-α. El infliximab se asocia con más reactivaciones del virus de la hepatitis B que etanercept o adalimumab108, probablemente debido a un aclaramiento más pronunciado del TNF-α con esta molécula, dado que infliximab se une tanto al TNF-α soluble como al ya unido al receptor y es, asimismo, citotóxico para las células que expresan TNF-α109.
En resumen, los nuevos agentes inmunosupresores no están exentos de toxicidad hepática, aunque esta reacción adversa es infrecuente. Una determinación del perfil hepático, la búsqueda de factores de riesgo de enfermedad hepática, incluída la determinación de autoanticuerpos, y la cuidadosa evaluación de la relación beneficio/riesgo deben preceder a la decisión terapéutica de instaurar tratamiento con estos fármacos98.

