





Durante la última década el conocimiento sobre la aterosclerosis ha experimentado una importante evolución. Considerada inicialmente una enfermedad por acumulación de colesterol, en la actualidad se asume que es una enfermedad inflamatoria resultado de una compleja interacción entre factores de riesgo, endotelio vascular y células sanguíneas, así como de los numerosos mensajes moleculares que se intercambian1,2.
Cuando el endotelio vascular se encuentra con ciertos productos bacterianos o factores de riesgo tan diversos como dislipemia, sustancias vasoconstrictoras implicadas en la hipertensión, productos de glucosilación asociados a la hiperglucemia o citocinas proinflamatorias derivadas de un exceso de tejido adiposo, las células aumentan la expresión de moléculas de adhesión, que promueven el contacto de los leucocitos de la sangre con la pared arterial. Estos leucocitos migran a través de la pared debido a la expresión de citocinas quimiotácticas reguladas por señales asociadas con factores de riesgo aterosclerótico. Una vez que residen en la íntima, los leucocitos, fundamentalmente monocitos y linfocitos T, se comunican con las células endoteliales y células musculares lisas (CML) de la pared del vaso. El mensaje intercelular que se produce depende de mediadores inflamatorios e inmunológicos, principalmente citocinas. Como consecuencia de este «fragor» inflamatorio, las CML migran desde la media hacia la íntima, proliferan y elaboran la matriz extracelular. En concierto con las células endoteliales y monocitos, las CML secretan metaloproteinasas, que modularán diversas funciones de las células vasculares, entre ellas la activación, proliferación, migración, neovascularización, remodelado, cicatrización y muerte celulares, así como degradación de la matriz extracelular. La rotura de una placa aterosclerótica vulnerable favorece la trombosis, causa de los síndromes clínicos cardiovasculares, como el infarto agudo de miocardio3,4.
El interés creciente por el papel de la inflamación en la aterosclerosis ha promovido el desarrollo de marcadores capaces de predecir acontecimientos cardiovasculares, como la proteína C reactiva, que emerge como un potente predictor del riesgo y añade valor predictivo al que proporcionan índices de riesgo vascular global, como el de Framingham5,6.
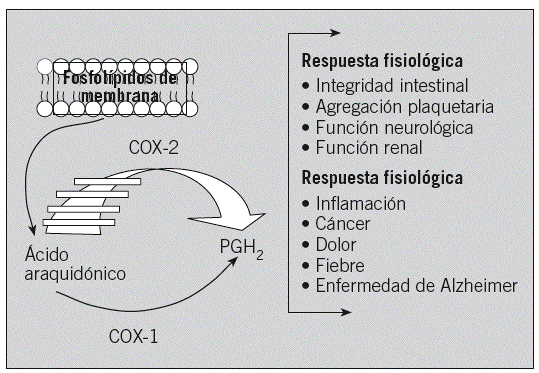
Los prostanoides, que incluyen prostaglandinas (PG) y tromboxanos (TX), pertenecen a un grupo de mediadores lipídicos derivados del ácido arquidónico vía ciclooxigenasas (COX). Participan en diversos procesos clínicos importantes, tales como la inflamación, trombosis, fiebre, reacciones alérgicas e inmunitarias, cáncer y enfermedad de Alzheimer7 (fig. 1).
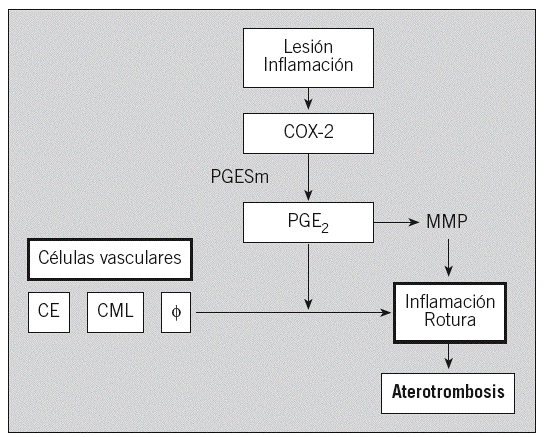
Fig. 1. Papel fisiopatológico de las ciclooxigenasas (COX). PGH2: prostaglandina H2 sintetasa.
La importancia de las COX en el ámbito cardiovascular se ve avalada por la eficacia de las dosis bajas de aspirina (inhibidor de la COX-1) en la prevención secundaria de enfermedades cardiovasculares8. Esta revisión se centra en el papel fisiológico y fisiopatológico de la COX-2 en el ámbito cardiovascular y se plantea si puede representar una nueva diana terapéutica en la aterosclerosis.
Ciclooxigenasas
En respuesta a la inflamación, los macrófagos secretan fosfolipasas, que catalizan la liberación de ácido araquidónico a partir de los fosfolípidos de membrana. Una vez liberado, el ácido araquidónico se convierte en diversos prostanoides por acción de las COX, también denominadas PGH-sintetasas9,10.
Se conoce desde la década de los años noventa la existencia de 2 isoformas de la COX: la COX-1 y la COX-2, codificadas por los cromosomas 9 y 1, respectivamente. El gen de COX-1, 9q32-33.3, posee 22 kilobases y 11 exones11. El gen de COX-2, 1q25.2-q25.3, posee 8,3 kilobases y 10 exones12. La activación transcripcional de COX-2 es inducida por diversos mediadores inflamatorios y mitogénicos, así como por factores hemodinámicos (shear stress)13.
La COX-1 y la COX-2 catalizan la primera etapa en la biosíntesis de PG al convertir el ácido araquidónico en el derivado PGH2, que isomerasas específicas metabolizarán en diferentes prostanoides, tales como PGE2, PGD2, PGF2a, PGI2 y TXA2, que ejercen diversas acciones biológicas en las proximidades de su lugar de síntesis activando receptores específicos en una gran variedad de células y tejidos9,10.
Asimismo, la COX-1 y COX-2 desempeñan un papel importante en la coagulación sanguínea, en la función renal y en el mantenimiento de la integridad gastrointestinal, pero también participan en procesos fisiopatológicos como la inflamación, la artritis y el cáncer. En general, la COX-1 es una enzima constitutiva que media funciones fisiológicas normales, mientras la COX-2 es una enzima inducible en respuesta a estímulos inflamatorios9,10. Frente a este concepto simplificado, emerge un nuevo paradigma que considera que la COX-2 puede ejercer efectos beneficiosos o nocivos dependiendo del tipo celular involucrado y el prostanoide generado: los leucocitos, las CML, las células endoteliales y las plaquetas expresan PGE-sintetasa y son capaces de generar el prostanoide proinflamatorio PGE2; las plaquetas expresan TX-sintetasa y elaboran el agente vasoconstrictor y protrombótico TXA2; las células endoteliales expresan PGI-sintetasa y sintetizan prostaciclina (PGI2), un prostanoide antitrombótico y con efectos vasodilatadores14,15. Además, se ha identificado recientemente una tercera isoforma de COX por procesamiento del ARN (splicing alternativo), la COX-3, cuyo papel fisiopatológico aún no se ha establecido16.
Ciclooxigenasas y función vascular
La COX-1 y la COX-2 desempeñan un papel importante en el mantenimiento de la homeostasis vascular. Ambas enzimas catalizan la formación de pequeñas cantidades de PGI2. En condiciones normales, el flujo laminar va a inducir la expresión de COX-2 en células endoteliales para generar PGI2 y estimular la liberación de óxido nítrico, que favorecen la integridad endotelial e inhiben la activación plaquetaria, con el consiguiente efecto antitrombótico17. En situaciones de inflamación vascular, las células endoteliales serían capaces de expresar concentraciones elevadas de COX-2, pero se produciría una situación de lesión vascular, con expresión de moléculas protrombóticas y reducción de la biodisponibilidad de óxido nítrico, además de una mayor generación de TXA2, con un efecto neto trombogénico en respuesta a estímulos proinflamatorios18-22.
Ciclooxigenasas en la aterosclerosis
Las COX pueden expresarse en todos los elementos celulares que participan en la aterosclerosis, tales como plaquetas, células entoteliales, monocitos, neutrófilos y linfocitos23. Diversos estímulos inflamatorios y metabólicos inducen la expresión de COX-2 por células vasculares, como el ligando CD4023, productos de glucosilación24, citocinas25, lipoproteínas de baja densidad26, estrés oxidativo27 y factores de crecimiento, como el factor de crecimiento vascular derivado del endotelio28.
En las arterias humanas, mientras que se ha observado ausencia o concentraciones indetectables de COX-2 en vasos normales29,30, existe un acentuado aumento en macrófagos y células endoteliales y, en menor medida, de CML en lesiones ateroscleróticas de carótida, aorta y coronarias31-33. Los macrófagos pueden sintetizar el derivado PGE2, un prostanoide aterogénico que induce la expresión de metaloproteinasas capaces de degradar la matriz extracelular34,35. En el «hombro» de la lesión aterosclerótica se ha demostrado la presencia de COX-2, que se colocaliza con metaloproteinasas en lesiones carotídeas sintomáticas36,37. También se ha observado un incremento de la PGE-sintetasa microsomal en células vasculares, que podría favorecer la biosíntesis de PGE2 y de metaloproteinasas dependientes de la COX-2 en lesiones ateroscleróticas38. Los receptores activados por proliferadores de los peroxisomas gamma, cuya expresión se encuentra aumentada en macrófagos de lesiones ateroscleróticas, regularían negativamente la expresión de COX-239.
Por consiguiente, la generación de prostanoides derivados de COX-2 promovería la aterosclerosis a través de diversos mecanismos, como quimiotaxis, permeabilidad vascular, migración, producción de citocinas proinflamatorias y proteólisis33,40; sin embargo, las vías específicas de señalización intracelular aún no se conocen con precisión.
Experimentalmente la deleción del receptor de prostaciclina (IP) acelera la aterogénesis en el ratón41 y la respuesta trombótica ante diversos estímulos42, mientras que la neutralización del receptor para el TX (TP) reduce la respuesta proliferativa ante la lesión vascular43.
Actividad de la ciclooxigenasa 2 monocitaria y riesgo cardiovascular
Hasta el momento son escasos los datos clínicos que evalúan la relación COX-2/PGE2 y el riesgo cardiovascular. En un estudio reciente en sujetos asintomáticos, nuestro grupo ha determinado la expresión de COX-2 y su actividad, medida como liberación monocitaria de PGE2, en relación con parámetros inflamatorios y de grosor íntima-media carotídeas, como expresión de aterosclerosis subclínica44. Se observó que la producción de PGE2 se incrementaba significativamente en relación con la presencia de factores tradicionales de riesgo vascular y que los sujetos en el tercil superior de PGE2 mostraban un incremento del grosor íntima-media en relación con terciles inferiores. La asociación entre ambos parámetros se mantenía en el análisis multivariante tras ajustar para los factores de riesgo (tabaquismo, diabetes, hipertensión e hipercolesterolemia) y parámetros inflamatorios (p. ej., proteína C reactiva), por lo que la PGE2 podría representar un factor independiente de riesgo cardiovascular y de aterosclerosis subclínica. Finalmente, se observó una relación significativa entre la expresión de COX-2 y la producción de PGE2, lo que apuntaría a un papel importante de la enzima en la biosíntesis del prostanoide proinflamatorio45. Estos resultados inducen a pensar que, al menos teóricamente, la COX-2 podría representar una nueva diana terapéutica en la aterosclerosis46,47.
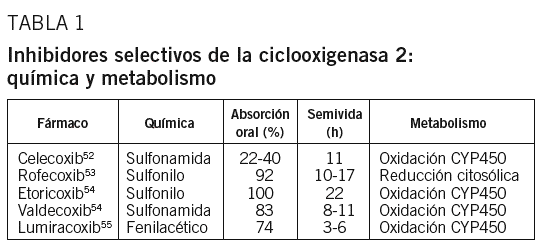
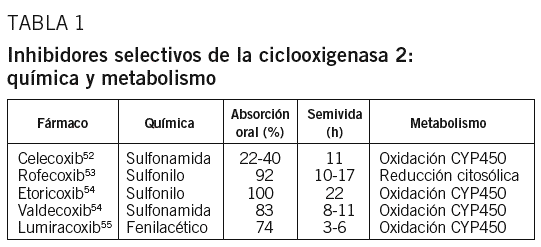
Ciclooxigenasa 2: ¿una diana terapéutica en la aterosclerosis?
El papel creciente de la inflamación en la patogenia de la aterosclerosis ha renovado el interés por la posibilidad de que agentes antiinflamatorios puedan ser eficaces en la prevención de episodios cardiovasculares de naturaleza aterotrombótica48-50. Por ejemplo, la aspirina, a través de su efecto inhibitorio de la agregación plaquetaria, reduce en un 25% la incidencia de infarto agudo de miocardio y otros episodios cardiovasculares51. Dicho efecto se ha relacionado con su acción inhibitoria de la COX-1 y de la generación de TX, sin afectar la generación vascular de PGI2, con el consiguiente efecto neto antitrombótico. Con la introducción de los inhibidores selectivos de la COX-2 o «coxib», tales como celecoxib, rofecoxib, etoricoxib, lumiracoxib y valdecoxib (tabla 1)52-55, fundamentalmente en el tratamiento de la artritis, para evitar los efectos gastrointestinales adversos de la aspirina y otros antiinflamatorios no esteroideos (AINE) clásicos (p. ej., naproxeno, ibuprofeno, diclofenaco, entre otros), se pensó que también podrían ejercer un efecto beneficioso sobre la aterosclerosis48-51. Experimentalmente existen evidencias de que la inhibición selectiva de la COX-2 puede ser beneficiosa sobre la aterosclerosis en algunos modelos56,57, pero no en otros58-60, y que puede inducir cardiotoxicidad61 o prevenirla62. También clínicamente existen estudios que demuestran un efecto beneficioso de los inhibidores selectivos de la COX-2 sobre la disfunción endotelial63,64 y la inflamación65, mientras que otros trabajos demuestran un incremento de la incidencia de episodios cardiovasculares relacionados con trombogenicidad y otros efectos adversos inducidos por estos fármacos66-68.
Inhibición de la ciclooxigenasa y complicaciones cardiovasculares
La explicación para la mayor incidencia de efectos adversos de naturaleza cardiovascular asociados con el empleo de inhibidores selectivos de la COX-2 podría estar en que estos compuestos bloquearían la generación de PGI2 sin inhibir la formación de TXA2, lo que daría como resultado un incremento de la activación y agregación plaquetarias que favorecería la trombosis y la presencia de episodios isquémicos66,67 (tabla 2). Además, la elevación de la presión sanguínea podría representar un factor de riesgo adicional68.

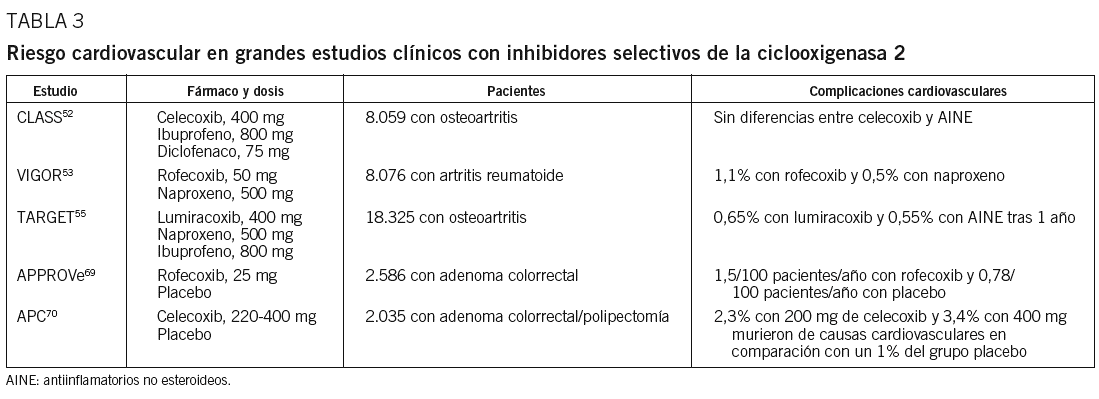
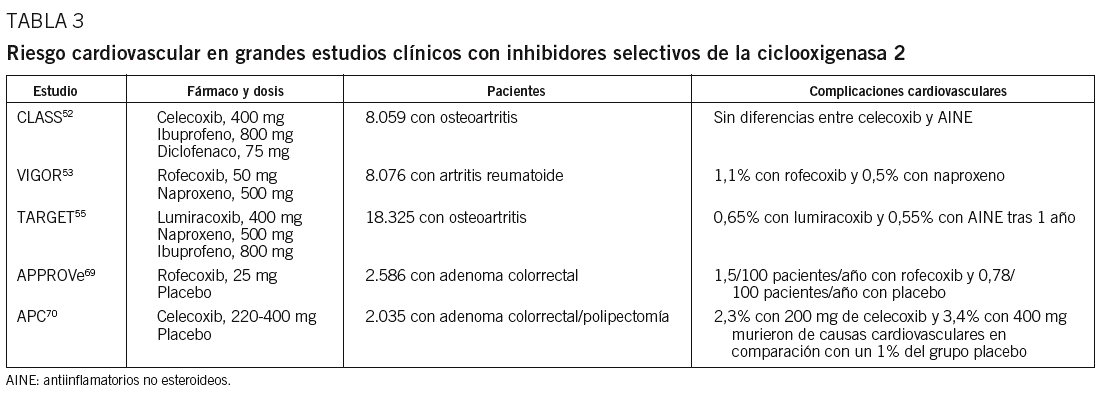
La evidencia clínica en la asociación de inhibidores selectivos de la COX-2 y episodios cardiovasculares procede de grandes estudios clínicos52,53,55,69,70 (tabla 3). En el estudio VIGOR53, en el que se comparaba el rofecoxib con el naproxeno en pacientes con osteortritis, se demostró una reducción significativa de episodios en este último grupo en relación con el tratado con inhibidores selectivos de la COX-2 (riesgo relativo de 2,38; intervalo de confianza del 95%, 1,39-4,05). El estudio Adenomatous Polyposis Prevention (APPROVe)69 se interrumpió tras observarse un incremento del riesgo de complicaciones aterotrombóticas después de 18 meses de tratamiento con rofecoxib, con retirada del fármaco por el laboratorio Merck en septiembre de 2004. En el estudio CLASS52, sin embargo, no se observó un aumento de episodios cardiovasculares en el grupo que recibió celecoxib en comparación con los tratados con diclofenaco e ibuprofeno, de forma similar a lo evidenciado en otros estudios realizados con este mismo fármaco y en metaanálisis71. En estudios con otros inhibidores selectivos de la COX-2, como el lumiracoxib, analizado en el estudio TARGET55, la incidencia de episodios cardiovasculares tras un año de seguimiento fue del 0,65% con el inhibidor selectivo de la COX-2 y del 0,55% con otros AINE. El estudio APC se interrumpió a los 33 meses de seguimiento al observarse un incremento de episodios cardiovasculares fatales y no fatales en el grupo que recibió celecoxib70. Estos resultados se han corroborado en metaanálisis que analizan la toxicidad cardiovascular con estos preparados72,73. En conjunto, si bien no todos los inhibidores selectivos de la COX-2 pueden mostrar un perfil de riesgo cardiovascular similar74, son numerosos los trabajos que indican un mayor riesgo de complicaciones vasculares en los pacientes tratados con inhibidores selectivos de la COX-2.
Conclusiones
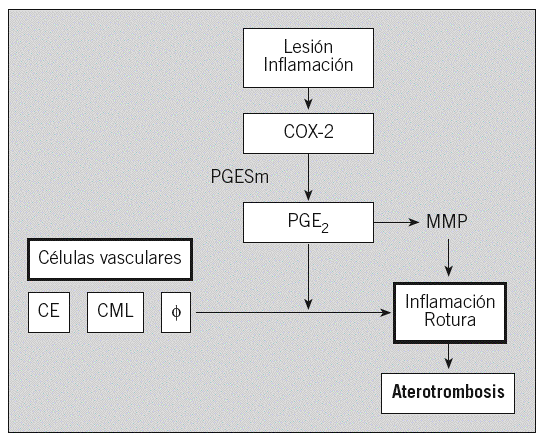
La COX-2 desempeña un papel importante en la fisiopatología cardiovascular dependiendo del tipo celular implicado y de la cantidad relativa de prostanoides que se generan. Estudios recientes de nuestro grupo indican que la vía COX-2/PGE2 puede ser asimismo de interés en la valoración del riesgo aterosclerótico en sujetos asintomáticos (fig. 2).
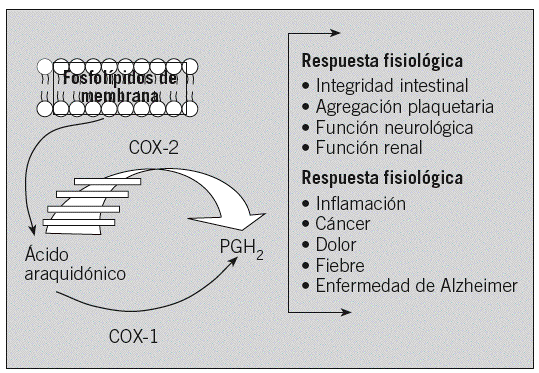
Fig. 2. Posible mecanismo de la vía ciclooxigenasa 2 (COX-2)/prostaglandina E2 (PGE2) en la aterosclerosis. CE: células endoteliales; CML: células del músculo liso; PGESm: prostanglandina E sintetasa microsómica; MMP: metaloproteinasas.
Sin embargo, por lo que respecta al empleo de inhibidores selectivos de la COX-2 en la prevención de la enfermedad aterosclerótica existe gran controversia en la actualidad54,75-77. Por ello, su uso debería restringirse, especialmente en las poblaciones de riesgo alto, ya que existen algunas cuestiones pendientes de resolver, tales como: ¿cuál es la magnitud del incremento del riesgo cardiovascular relacionado con los inhibidores selectivos de la COX-2?, ¿varía el riesgo según el tipo de fármaco?, ¿es similar el riesgo cardiovascular al de AINE que no inhiben selectivamente la COX-2? Si las circunstancias clínicas aconsejan el uso de inhibidores selectivos de la COX-2 durante un período de tiempo prolongado, la administración de aspirina (80-100 mg/ día) puede ser útil en la prevención de complicaciones futuras. En todo caso, se recomienda reducir la duración del tratamiento y realizar un seguimiento atento de la presión arterial, del desarrollo de edemas, de la función renal y de la hemorragia gastrointestinal68.
Es probable que nuevas moléculas sin las limitaciones actuales de los inhibidores selectivos de la COX-2 puedan ser de interés, como los agentes capaces de inhibir simultáneamente las vías de la COX y la lipooxigenasa78. Asimismo, la inhibición de la PGE2, metabolito proinflamatorio derivado de la COX-2, podría constituir otra opción, que evitaría las consecuencias de la inhibición completa de la vía COX-238.
En conclusión, aún es prematuro considerar la COX-2 una diana terapéutica en la aterosclerosis, debido a los riesgos potenciales sobre la incidencia de episodios cardiovasculares asociados al empleo de inhibidores selectivos de la COX-2. Estudios clínicos en curso pueden proporcionar la respuesta sobre el efecto neto de la inhibición selectiva de la COX-2 sobre el riesgo cardiovascular.
