




Durante la diabetes mellitus tipo 2 se produce un aumento en la expresión del transportador proximal de glucosa SGLT2. Este aumento indebido de la recuperación de glucosa filtrada desde la orina hacia el túbulo proximal y, posteriormente, hacia el torrente sanguíneo tiene 3 repercusiones directas sobre el pronóstico del paciente con diabetes mellitus tipo 2: a) aumento de la carga diaria de glucosa al elevar el umbral de reabsorción. Como consecuencia aumentan los requerimientos de antidiabéticos orales y de insulina. Este aumento es progresivo a lo largo de la enfermedad y va a la par con el aumento de masa renal (hipertrofia renal); b) la mayor reabsorción de glucosa hace que la glucosuria sea menor de la correspondiente al valor de glucemia, disminuyendo el estímulo sobre el “feed-back” tubuloglomerular de la nefrona distal. Como resultado, no se contrarresta la vasodilatación glomerular causada por la hiperglucemia, perpetuándose la hiperfiltración glomerular, y c) el exceso de glucosa transportado a la célula del túbulo proximal modifica el estado redox de esta, aumentando la producción local de productos con capacidad glucosilativa y activando la producción local de mediadores proliferativos proinflamatorios y profibróticos. Estos mediadores son responsables del daño directo por radicales libres a la célula tubular proximal, del aumento de expresión de SGLT2, del aumento de producción de colágeno IV y de matriz extracelular, y de la activación de monocitos-macrófagos con capacidad lesiva endotelial. La utilización de inhibidores del transporte proximal de glucosa SGLT2 no solo reduce la reinyección de la glucosa ya filtrada desde la orina hacia la sangre, mejorando el control metabólico de la diabetes, sino que además restaura el “feed-back” tubuloglomerular al aumentar la glucosuria y el flujo urinario distal. Pero el efecto más notable se debe a la inhibición de la entrada de glucosa en la célula tubular proximal. La glucosuria es tóxica para el riñón: lo es para las células capaces de transportar glucosa, es decir, las células proximales dotadas de SGLT2. En modelos animales, al inhibir SGLT2 se reduce la producción local de radicales de oxígeno, disminuye la formación de matriz mesangial y de colágeno IV, hay menor infiltración glomerular por células inflamatorias y se reduce la arteriosclerosis dependiente de monocito-macrófago. En clínica humana, los iSGLT2 han mostrado su capacidad para reducir el daño renal y el riesgo cardiovascular de los pacientes con diabetes mellitus tipo 2.
In DM2, there is increased expression of the proximal glucose transporter SGLT2. The increased glucose reabsorption from the urine to the proximal tubule and subsequently to the bloodstream, has three direct effects on the prognosis of patients with DM2: a) it increases the daily glucose load by raising the renal threshold for glucose, thus augmenting requirements for oral antidiabetics and insulin. This progressive increase occurs throughout the course of the disease and in parallel with the increase in renal mass (renal hypertrophy); b) because of the greater glucose reabsorption, glycosuria is lower than the level corresponding to glycaemia, decreasing the stimulus on the tubuloglomerular feedback system of the distal nephron. As a result, the glomerular vasodilation caused by hyperglycaemia is not arrested, maintaining glomerular hyperfiltration, and c) the excess glucose transported to the proximal tubular cells modifies their redox status, increasing local production of glycosylating products and activating local production of proinflammatory and profibrotic proliferative mediators. These mediators are responsible for the direct free radical damage to proximal tubular cells, for increased SGLT2 expression, increased production of collagen IV and extracellular matrix, and activation of monocyte/macrophages able to cause endothelial injury. The use of SGLT2 inhibitors not only reduces the reabsorption of glucose from the glomerular filtrate back into the circulationthus improving metabolic control in diabetesbut also restores tubuloglomerular feedback by increasing glycosuria and distal urinary flow. However, the most notable effect is due to inhibition of glucose entry to the proximal tubular cells. Glycosuria is toxic to the kidney: it harms glucosetransporting cells, that is, the proximal cells, which contain SGLT2. In animal models, SGLT2 inhibition reduces local production of oxygen-free radicals, the formation of mesangial matrix and collagen IV, glomerular infiltration by inflammatory cells and monocyte/macrophage-dependent arteriosclerosis. In humans, SGLT2 have a demonstrated ability to reduce renal injury and cardiovascular risk in patients with type 2 diabetes mellitus.
Los inhibidores del SGLT2 (cotransportador sodio-glucosa 2) son fármacos capaces de inhibir en un determinado porcentaje, próximo a un 50%, la reabsorción renal de glucosa, impidiendo el paso de esta desde la luz del túbulo proximal renal al interior celular1,2. El resultado de dicha inhibición es un aumento en la cantidad de sodio y glucosa, que alcanzan segmentos más distales de la nefrona. El sodio resulta parcialmente atrapado en el asa de Henle y en el túbulo distal, que funcionan según el principio de carga: si aumenta la llegada de sodio aumenta la reabsorción de este. Pero la glucosa no tiene transportadores efectivos en el resto de la nefrona, por lo que se excreta en la orina. El volumen de diuresis que arrastra depende de la presencia o ausencia de hormona antidiurética y de la concentración de solutos a nivel del intersticio medular y, en general, aumenta entre 500 y 2.000 ml más al día3.
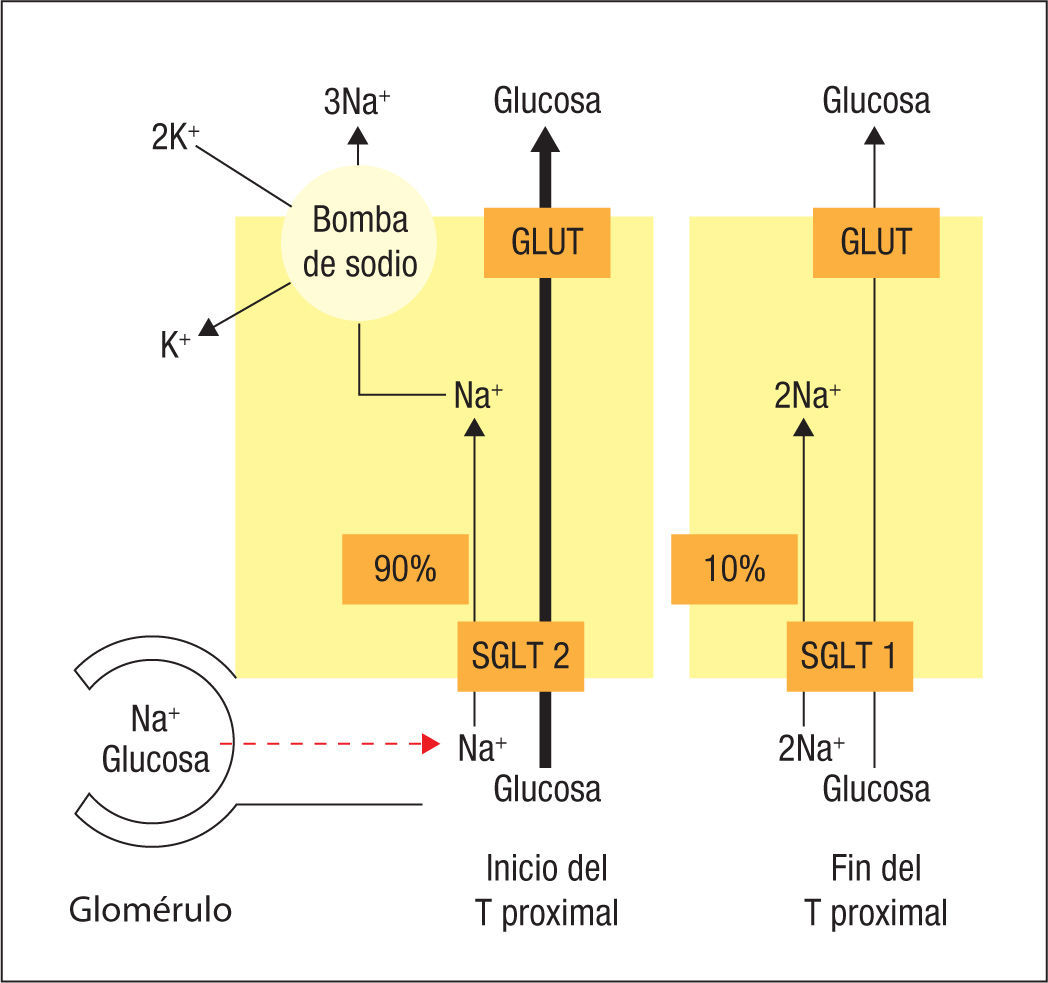
La reabsorción proximal de glucosa está muy aumentada en la diabetes mellitus tipo 2La glucosa pertenece a un grupo de sustancias cuyo manejo renal incluye una filtración importante, seguida de la recuperación prácticamente completa mediante reabsorción tubular4,5. En un paciente con un filtrado glomerular de 180 l/día (120 ml/min) y una glucemia de 100 mg/dl (1 g/l), la cantidad de glucosa filtrada diariamente es de 180 g/día. Nada de esta glucosa alcanza la orina, porque es reabsorbida en su totalidad por el túbulo proximal: un 90% a través del SGLT2, de baja afinidad pero alta capacidad, capaz de reabsorber una molécula de glucosa por cada molécula de sodio, y un 10% a través del SGLT1, localizado algo más adelante en el túbulo proximal, de alta afinidad y baja capacidad, que reabsorbe 1 molécula de glucosa por cada 2 moléculas de sodio5–7 (fig. 1). En ambos casos, la glucosa pasa de la célula al capilar a través de uniportadores de glucosa tipo GLUT.
 y el 10% en el segmento S3 (final). Los transportadores apicales cotransportan 1 molécula de glucosa con 1 (SGLT2) o con 2 moléculas de sodio (SGLT1). Los transportadores basolaterales son uniportadores de la familia GLUT. El transporte glucosilado es activado por una bomba de sodio basolateral 3Na+x 2K+ (referencias 1 y 5). SGLT1: sodium glucose linked transport type 1; SGLT2: sodium glucose linked transport type 2.")
Mecanismos de reabsorción renal de glucosa. La totalidad de la glucosa reabsorbida es transportada en el túbulo proximal: el 90% lo es en el segmento S1 (inicio) y el 10% en el segmento S3 (final). Los transportadores apicales cotransportan 1 molécula de glucosa con 1 (SGLT2) o con 2 moléculas de sodio (SGLT1). Los transportadores basolaterales son uniportadores de la familia GLUT. El transporte glucosilado es activado por una bomba de sodio basolateral 3Na+x 2K+ (referencias 1 y 5). SGLT1: sodium glucose linked transport type 1; SGLT2: sodium glucose linked transport type 2.
Cuando aumenta transitoriamente la glucemia, aumenta la carga filtrada de glucosa (carga filtrada g/día = concentración plasmática g/l x filtrado glomerular l/día), por lo que el sistema en tándem de SLGT2 y SLGT1 se acaba saturando y comienza a aparecer glucosuria. La glucosuria se inicia para valores de glucemia plasmática en torno a 1,8-2 g/l, y es un mecanismo regulador para devolver la glucemia a sus valores normales4.
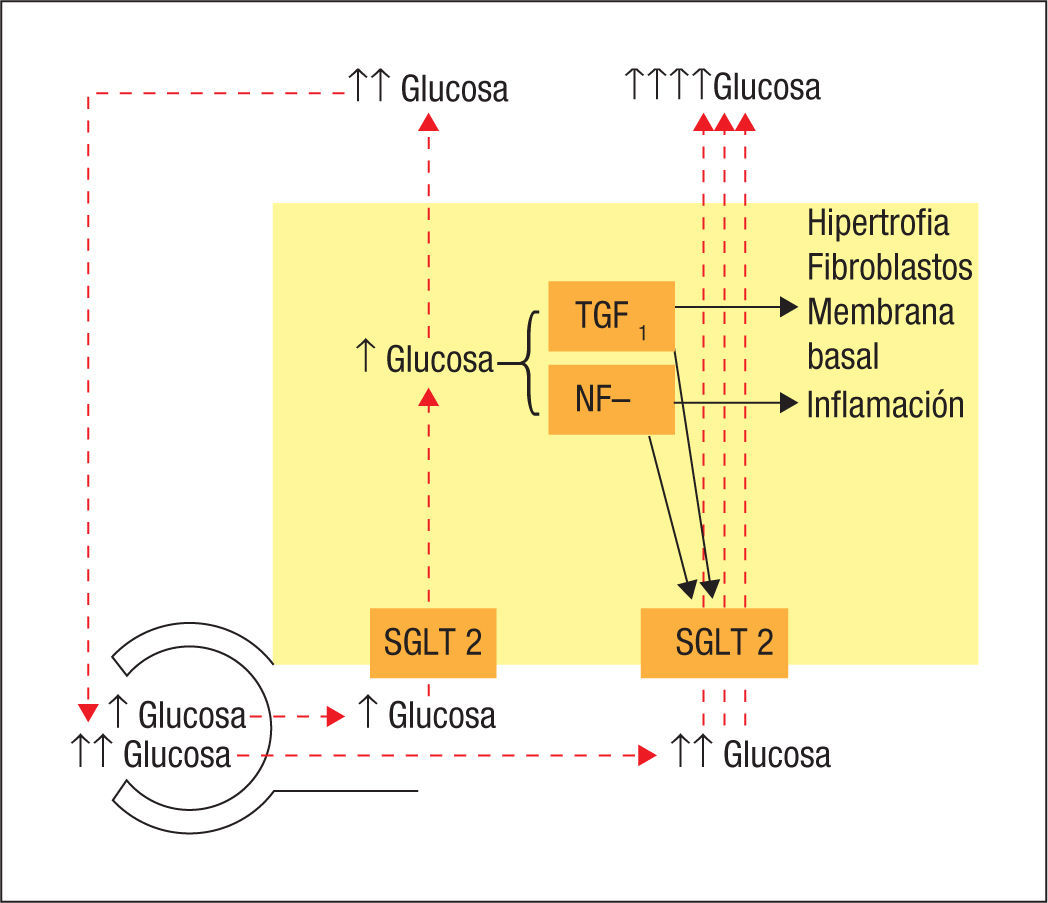
Sin embargo, cuando la carga filtrada de glucosa se mantiene elevada y la concentración de glucosa en el túbulo es elevada, el aumento de glucosa transportada hacia el interior de la célula tubular proximal estimula la producción local del factor de crecimiento transformador beta (TGFp)8, interleucina (IL)-6 y factor de crecimiento tumoral alfa (TNFa)9, cuyas concentraciones locales aumentan de modo mantenido (fig. 2). Estos mediadores celulares se han implicado en la respuesta proliferativa renal y vascular y son, en buena medida, responsables de la fibrogénesis y del ensanchamiento de la membrana basal de las distintas estructuras renales. Pero además son capaces de aumentar la síntesis local de SGLT2, aumentando el transporte de glucosa hacia el interior de la célula y desde ahí hacia la circulación8,9.

El aumento en la glucosa filtrada que acompaña a la hiperglucemia causa un aumento en la glucosa intracelular que determina en menos de 48 h el aumento de actividad de SGLT28–10. La activación de TGFp y NF-κB se ha implicado en este proceso. Los efectos autocrinos de estos mediadores son responsables del aumento en la expresión y función de SGLT2. Los efectos paracrinos implican activación a distancia de células inflamatorias, proliferación celular y fibrosis local8,9. NF-κB: factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas; SGLT2: cotransportador sodio-glucosa 2; TGFβ: factor de crecimiento transformador beta.
La capacidad celular de transporte de glucosa del túbulo proximal es al menos 4 veces mayor de lo normal10, lo que implica que el riñón del paciente con diabetes mellitus tipo 2 (DM2) está reinyectando hacia la circulación sistémica no menos de medio kilo diario de glucosa.
En presencia de DM2 con hiperglucemia mantenida se produce glucosuria, pero dicha glucosuria es inferior a la que debería observarse para ese nivel de hiperglucemia.
Esta hiperactividad de SGLT2 en la DM2 determina 3 conflictos:
- 1.
Hay una sobrecarga de glucosa al metabolismo intermedio de la célula proximal.
- 2.
Hay una reducción relativa en la eliminación urinaria de glucosa.
- 3.
Hay una oferta de glucosa desde el riñón a la economía inapropiadamente alta.
Cada uno de estos 3 conflictos podría tener repercusiones en la hemodinámica del sujeto, en su supervivencia renal y en el riesgo cardiovascular de este.
La comprensión de los efectos globales de los inhibidores de SGLT2 (iSGLT2) requiere considerar su impacto en todos los aspectos descritos.
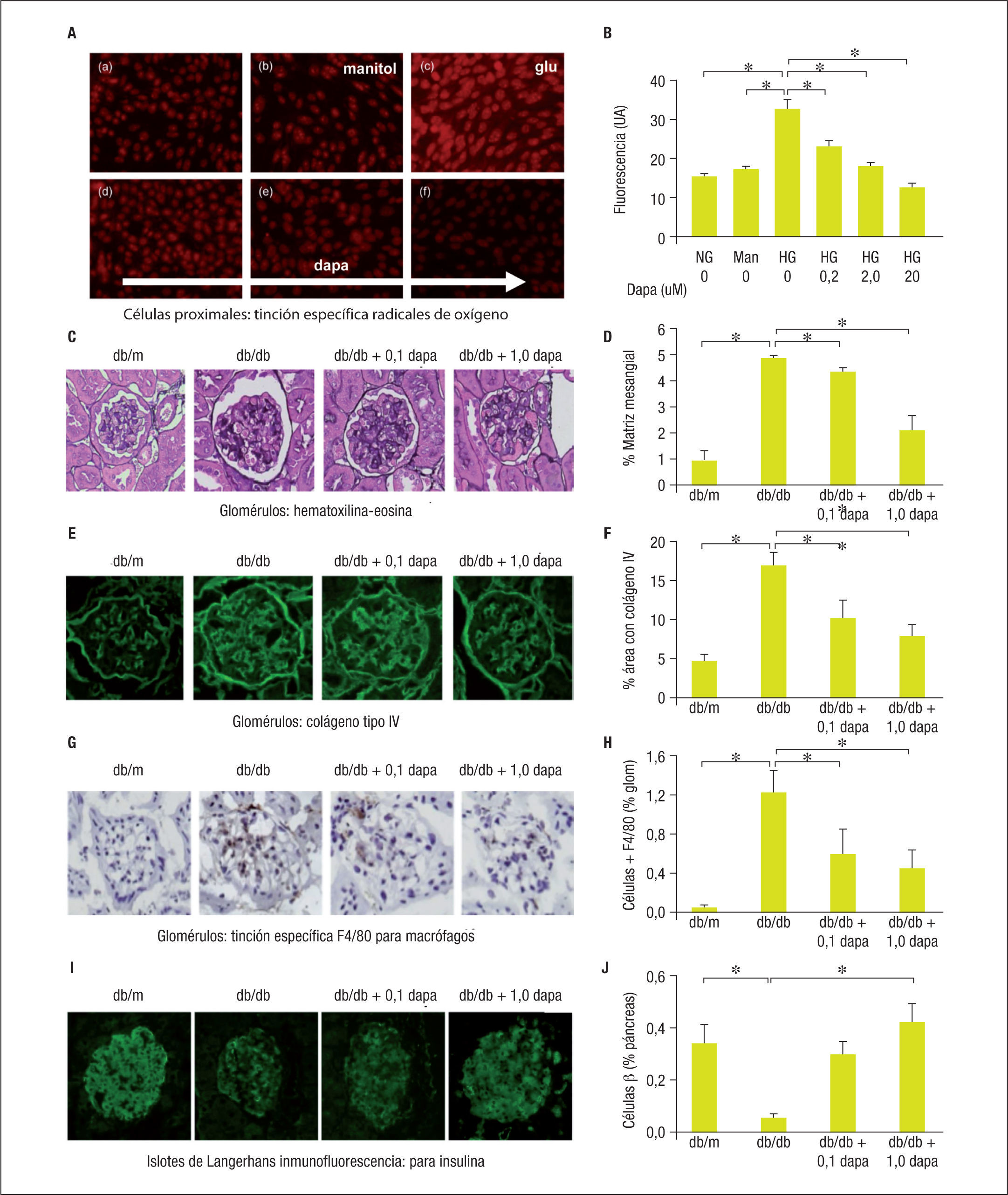
Sobrecarga de glucosa a la célula renal: oxidación, inflamación y proliferaciónLa glucosuria es tóxica para el riñón. Esta afirmación, conocida desde hace mucho tiempo, necesita ser matizada: es tóxica para las células renales que utilizan glucosa, es decir, es tóxica para las células del túbulo proximal. El resto de la nefrona apenas tiene transporte de glucosa, siempre a través de uniportadores tipo GLUT. En un modelo murino de DM2, la presencia de glucosa en la vertiente apical de las células del túbulo proximal aumenta la producción celular de radicales solubles de oxígeno (ROS)11 (fig. 3A). La sustitución de glucosa por otro azúcar no metabolizable, como el manitol, no causa daño celular11. Al inhibir la función de SGLT2 se aumenta la glucosuria distal protegiéndose la única célula capaz de ser lesionada por la glucosuria, la célula del túbulo proximal11 (fig. 3B).
 obtenido por mutación y selección de mutantes homocigotos para el receptor de leptina, los animales desarrollan un fenotipo de hiperfagia, obesidad, hiperinsulinismo e hiperglucemia con datos de resistencia a la insulina11. Dapa: dapagliflozina; db/m: heterocigoto, no diabético; db/ db homocigoto diabético. A) Detección de radicales solubles de oxígeno (ROS) mediante tinción con dihidroetidio (DHE) (Molecular Probes, OR, USA). B) Cuantificación de fluorescencia obtenida en A, correspondiente a la presencia de ROS. C) Histología renal, hematoxilina-eosina glomérulo (x400). D) Índice de matriz mesangial estimado mediante el software B102ERO (Keyence). E) Expresión de colágeno tipo IV a nivel glomerular. Doble inmunotinción con anticuerpo específico anticolágeno IV de conejo (Millipore, Temecula CA; USA) y cuantificado como en D. G) Infiltración de monocitos-macrófagos: tinción de inmunoperoxidasa con anticuerpos monoclonales antimonocito-macrófago C Abram, F4/80. H) Cuantificación de células positivas para la inmunotinción de peroxidasa. I) Islotes de Langerhans. Detección de células β mediante anticuerpos antiinsulina de cobaya (Abcam, Cambridge, UK) y anticuerpos de cabra anti-IgG de cobaya (Invitrogen). J) Cuantificación del área del islote ocupado por células β como en D11.")
En un modelo murino de diabetes mellitus tipo 2 (DM2) obtenido por mutación y selección de mutantes homocigotos para el receptor de leptina, los animales desarrollan un fenotipo de hiperfagia, obesidad, hiperinsulinismo e hiperglucemia con datos de resistencia a la insulina11. Dapa: dapagliflozina; db/m: heterocigoto, no diabético; db/ db homocigoto diabético. A) Detección de radicales solubles de oxígeno (ROS) mediante tinción con dihidroetidio (DHE) (Molecular Probes, OR, USA). B) Cuantificación de fluorescencia obtenida en A, correspondiente a la presencia de ROS. C) Histología renal, hematoxilina-eosina glomérulo (x400). D) Índice de matriz mesangial estimado mediante el software B102ERO (Keyence). E) Expresión de colágeno tipo IV a nivel glomerular. Doble inmunotinción con anticuerpo específico anticolágeno IV de conejo (Millipore, Temecula CA; USA) y cuantificado como en D. G) Infiltración de monocitos-macrófagos: tinción de inmunoperoxidasa con anticuerpos monoclonales antimonocito-macrófago C Abram, F4/80. H) Cuantificación de células positivas para la inmunotinción de peroxidasa. I) Islotes de Langerhans. Detección de células β mediante anticuerpos antiinsulina de cobaya (Abcam, Cambridge, UK) y anticuerpos de cabra anti-IgG de cobaya (Invitrogen). J) Cuantificación del área del islote ocupado por células β como en D11.
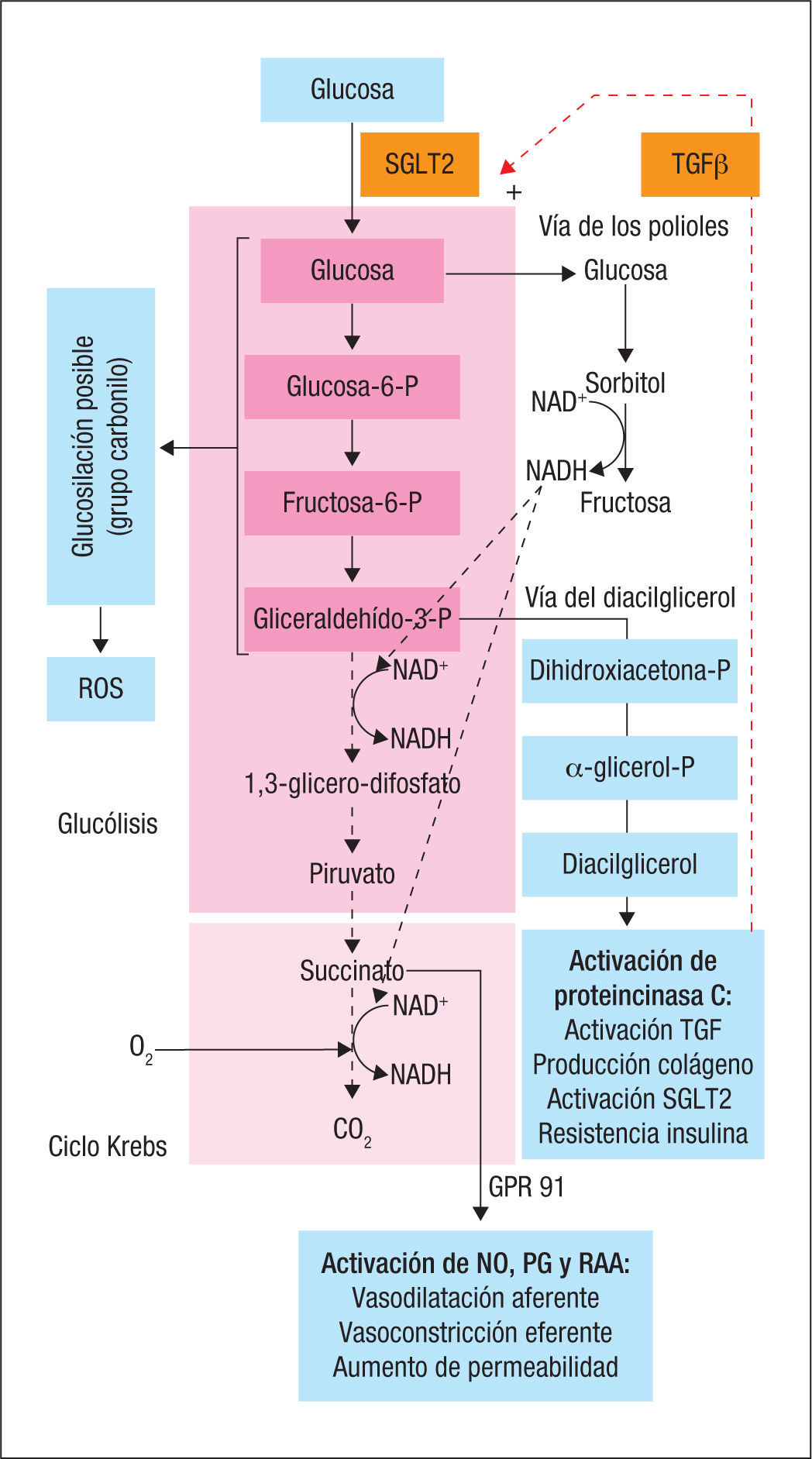
Al igual que ocurre en otros tejidos con escasa actividad glucolítica (riñón, retina, cristalino, corazón, sistema nervioso central), el exceso de glucosa satura rápidamente la capacidad de las vías glucolíticas del túbulo proximal, derivándose hacia vías secundarias como la de los polioles12 (fig. 4), transformando la glucosa en productos no metabolizables. Pero esta vía genera una cantidad considerable de NADH (equivalentes de reducción); el acúmulo de NADH dificulta las reacciones redox, cuyo producto final es también NADH13. Así, la oxidación de las triosas se bloquea14, y se bloquea también la oxidación de algunos intermediarios del ciclo de Krebs como el succinato. Los efectos de estos 2 bloqueos son sorprendentes e inesperados.

El bloqueo de la oxidación de triosas (gliceraldehído 3P) causa la acumulación de todos los intermediarios de la glucólisis, todos ellos con capacidad glucosilativa15. La glucosilación intracelular de proteí nas que se produce es mucho mayor que la extracelular16. Y los productos de glucosilación avanzados (AGE) resultantes de esta actividad glucosilativa intracelular generan radicales libres de oxígeno, activación de factores de transcripción nuclear proinflamatorios, como el NF-κp, proliferación celular, expresión de IL-6 y PAI-1 (inhibidor del activador del plasminógeno) y proteínas de matiz extracelular17. Estos son los efectos auto y paracrinos del aumento en la oferta de glucosa a la célula tubular renal, verdaderos ejecutores renales del daño por exceso de transporte renal de glucosa. Otros tipos celulares con sistemas de transporte de glucosa no acoplado al sodio (GLUT2), como el hepatocito o la célula beta del islote de Langerhans, muestran efectos similares en la producción intracelular de AGE debido al acúmulo de metabolitos intermediarios de la glucólisis, mucho más reactivos que la propia glucosa. En ambos tejidos se ha descrito la acumulación de AGE en las histonas de los núcleos celulares, donde interfieren con la proliferación celular y la expresión génica18.
El gliceraldehído 3-P deriva a dehidroxiacetona fosfato y así las triosas procedentes del exceso de oxidación de glucosa entran en la vía del diacilglicerol, segundo mensajero capaz de activar la protein-cinasa (PKC) en la célula renal, en la endotelial y en la retina19. La activación de PKC supone un evento crucial en el daño vascular, retiniano y renal asociado a la DM220.
- –
La PCK renal, activada por la hiperglucemia y el exceso de oferta de glucosa a la célula tubular a través de SGLT2, favorece la señalización de insulina a través de vías independientes de los sustratos del receptor de insulina, favoreciendo las acciones proliferativas, desdi-ferenciadoras y de supervivencia de la insulina, e interfiriendo con las vías de señalización metabólica, es decir, empeorando la resistencia a la insulina.
- –
La PKC activa TGFp, mediador clave en la hipertrofia renal y en la síntesis de colágeno. Además, TGFp aumenta la síntesis y la actividad del propio SGLT2, cerrando un círculo nocivo que aumenta la lesión glucosilativa renal21.
- –
La PKC estimula la síntesis de TNFa y de otros medidores inflamatorios, probablemente causando un efecto paracrino y, tal vez, a distancia sobre otros órganos.
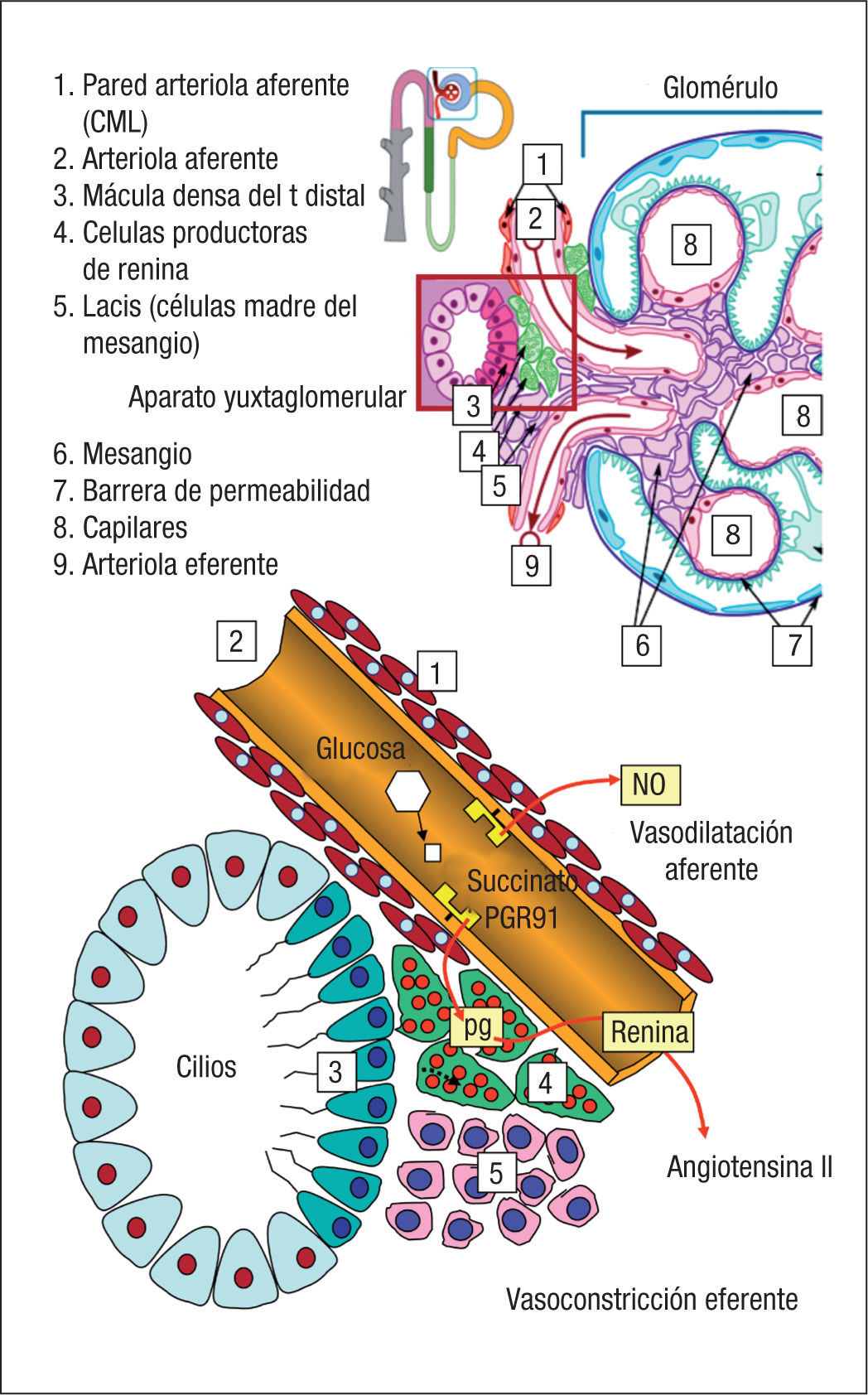
El bloqueo en la oxidación del succinato aumenta los valores locales de este. Se ha propuesto que la elevación del succinato circulante durante la hiperglucemia puede contribuir de modo decisivo a la vasodilatación de la arteriola aferente implicada en la hiperfiltración inducida por la hiperglucemia22,23 (fig. 5).
, prostaglandinas y renina22,23.")
La generación intracelular de AGE es responsable de que la célula tubular renal sometida a altas concentraciones de glucosa se transforme en una fuente de lesión oxidativa.
La activación de NF-κp de la célula renal sometida a altas concentraciones de glucosa transforma dicha célula en una fuente local de inflamación24 y, finalmente, la activación de PKC y TGFp provoca la activación local de mecanismos profibróticos, con aumento del colágeno tipo IV y proliferación mesangial21.
En un modelo experimental de DM2, la simple inhibición farmacológica del transporte renal de glucosa SGLT2 redujo la infiltración inflamatoria del glomérulo, la síntesis y depósito de colágeno IV a nivel glomerular y peritubular, y la disminución de la matriz mesangial11, mostrando el papel iniciador del transporte de glucosa por la célula tubular en varios de los fenómenos clásicamente asociados a la nefropatía diabética (fig. 3C-F).
Este efecto es aditivo al observado con la inhibición del eje renina-angiotensina-aldosterona25.
No es la primera vez que se observa el papel paracrino que juega la lesión de la célula tubular renal en la quimiotaxis de células inflamatorias hacia el riñón. Recientemente nuestro grupo ha mostrado que la lesión específica del túbulo proximal con un agente nefrotóxico provoca la activación de NF-κp26 y la infiltración inmediata de la zona lesionada por células monocito-macrófago27. Si se inhibe la apoptosis inducida por el nefrotóxico, no se activa NF-κβ y deja de producirse la infiltración inflamatoria27.
Por último, la retroalimentación a través de la activación de PKC, NF-κβ, TGFβ, IL-6 y TNFα, con la activación y aumento de expresión consiguiente de SGLT2, determina un aumento progresivo en la “reinyección” de glucosa desde el túbulo hacia el torrente circulatorio, ya que es SGLT2 y no GLUT2 el factor limitante del transporte vectorial de glucosa en el túbulo proximal.
Reducción relativa en la glucosuria: papel sobre la hiperfiltraciónEl aumento en la reabsorción renal de glucosa observado en la DM2 hace que el flujo distal de orina, aun estando aumentado con respecto a un paciente sin diabetes, sea menor de lo que correspondería para el nivel de glucosuria.
El flujo distal es percibido a nivel de la mácula densa, una parte especializada del túbulo distal, que se encuentra en directa aposición con las arteriolas aferente y eferente del glomérulo, con las células productoras de renina del aparato yuxtaglomerular y con las células del mesangio (“lacis”) (fig. 5).
A este nivel, el flujo de orina —percibido a través del movimiento pasivo que provoca en los cilios de la mácula densa— ejerce un efecto directo sobre la arteriola aferente, cuyo tono regula. Cuando aumenta el flujo distal se eleva el tono de la arteriola aferente, lo que resulta en su vasoconstricción. Cuando el flujo distal es menor de lo debido, el tono vascular aferente se relaja y hay vasodilatación. Este mecanismo está mediado por receptores de adenosina A1 presentes en la pared de la arteriola aferente, y se conoce como “feed-back” tubuloglomerular.
El aumento progresivo en la reabsorción proximal de glucosa y sodio a través de SGLT2 condiciona un flujo distal menor del esperado y una reducción en el “feed-back” tubuloglomerular. Probablemente, esta situación no tendría mayor relevancia, si no fuese porque la hiperglucemia del paciente con DM2 condiciona por sí misma un aumento basal en la filtración glomerular.
En 2008, Toma et al mostraron por primera vez que la hiperglucemia causa vasodilatación aferente y activación directa del eje renina-angiotensina23. En presencia de hiperglucemia aumentan los valores circulantes de succinato (fig. 4), que se detectan por un receptor endotelial situado a nivel de la arteriola aferente. La activación de dicho receptor genera un aumento en la síntesis local de óxido nítrico (NO), vasodilatación de la arteriola aferente e hiperfiltración23.
La hiperfiltración es detectable incluso en pacientes no diagnosticados aún de DM2 con intolerancia a carbohidratos28.
Cuando se produce una sobrecarga puntual de glucosa, la hiper-glucemia causa un aumento en la filtración glomerular. Esta hiperfiltración produce un aumento de la carga filtrada de glucosa, del flujo distal de orina y de la excreción urinaria de glucosa. La activación del “feed-back” tubuloglomerular correspondiente sirve para minimizar la hiperfiltración hasta que la glucemia se normaliza.
Pero en el paciente con DM2, la hiperfunción de SGLT2 lo trastoca todo, ya que no se produce el “feed-back” esperado, y la hiperfiltración desencadenada por la hiperglucemia no se compensa.
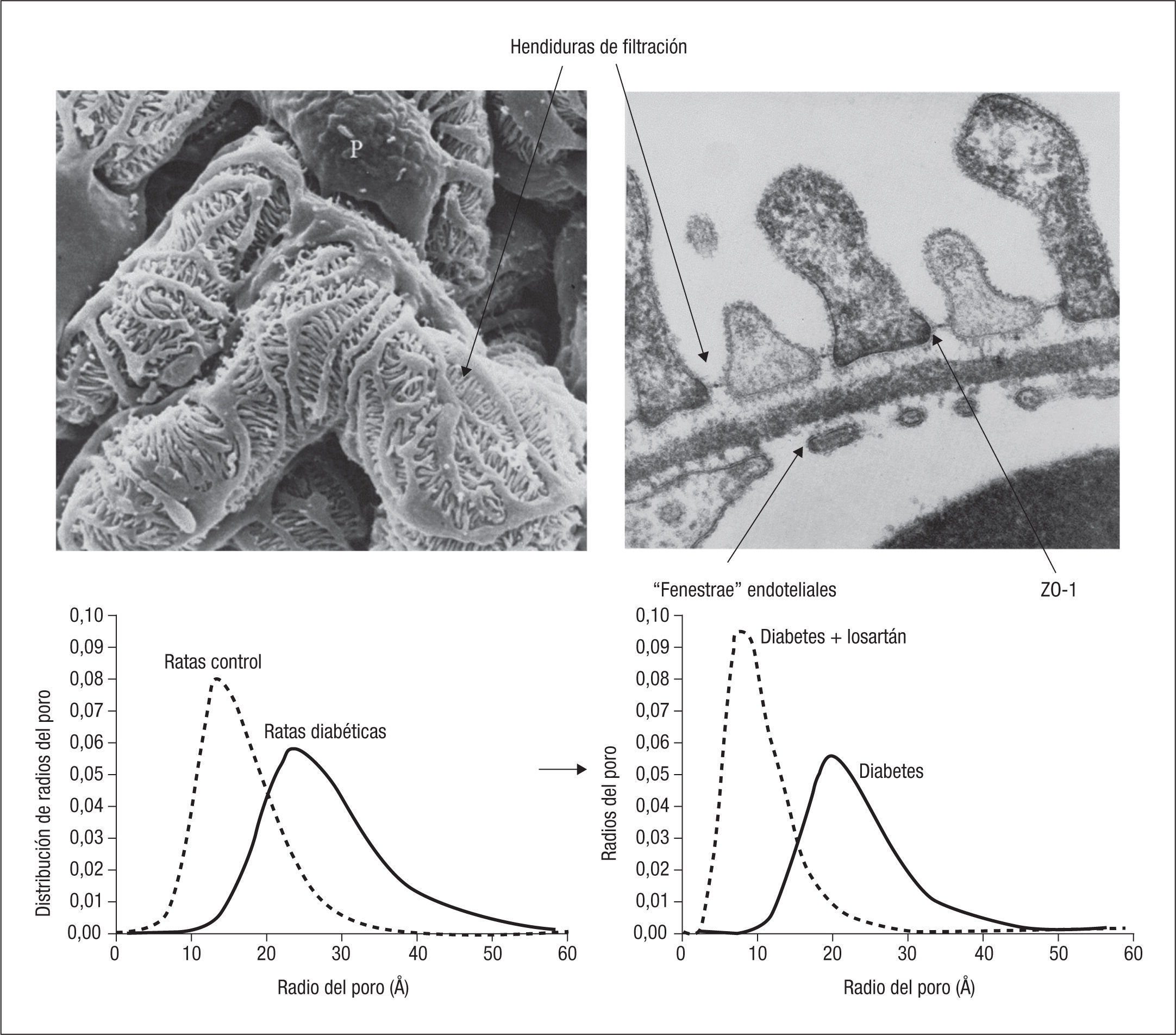
Cuando hay hiperfiltración, la distensión de la pared de los capilares aumenta el diámetro de las hendiduras entre los pedicelos de los podocitos (fig. 6), lo que facilita la fuga de albúmina. En animales de experimentación sometidos a la inducción de una DM1, se ha podido determinar el tamaño de dichas hendiduras en situación basal y tras la inducción de diabetes. Si se baja la presión intraglomerular de los animales diabéticos con un inhibidor de la enzima de conversión de la angiotensina (IECA), el diámetro de las hendiduras vuelve a la normalidad, al tiempo que se reduce la albuminuria29,30.
 en glomérulos de animales control, animales diabéticos y animales diabéticos tratados con losartán29,30.")
Fotografía microscópica de los capilares de un glomérulo de roedor cubiertos por los podocitos. Detalle de la barrera de permeabilidad, con las “menestras” endoteliales y las zónulas occludens del podocito. Distribución de frecuencias del tamaño de los poros (radios) en glomérulos de animales control, animales diabéticos y animales diabéticos tratados con losartán29,30.
Hasta el momento, la forma de evitar o reducir la hiperfiltración era el control metabólico estricto para evitar la hiperglucemia mantenida, y el uso de IECA o antagonistas de los receptores de la angiotensina II para dilatar la arteriola eferente y reducir la presión intraglomerular.
La disponibilidad de inhibidores del transportador SGLT2 añade una herramienta nueva para evitar la hiperfiltración de los primeros estadios de la nefropatía diabética.
La primera prueba de que la inhibición del transporte renal de glucosa a través de SGLT2 podía corregir la hiperfiltración de la DM fue publicada por Cherney et al en 201431. Mostraron que la hiperfiltración observada en la DM1, tanto en condiciones de clamp euglucémico como de clamp hiperglucémico, retornaba a la normalidad al usar un iSGLT-2. Y esta normalización de la hiperfiitración estaba mediada por una reducción en la producción de NO31 (la indicación de uso de la clase iSGLT-2 no está autorizada en diabetes mellitus tipo 1).
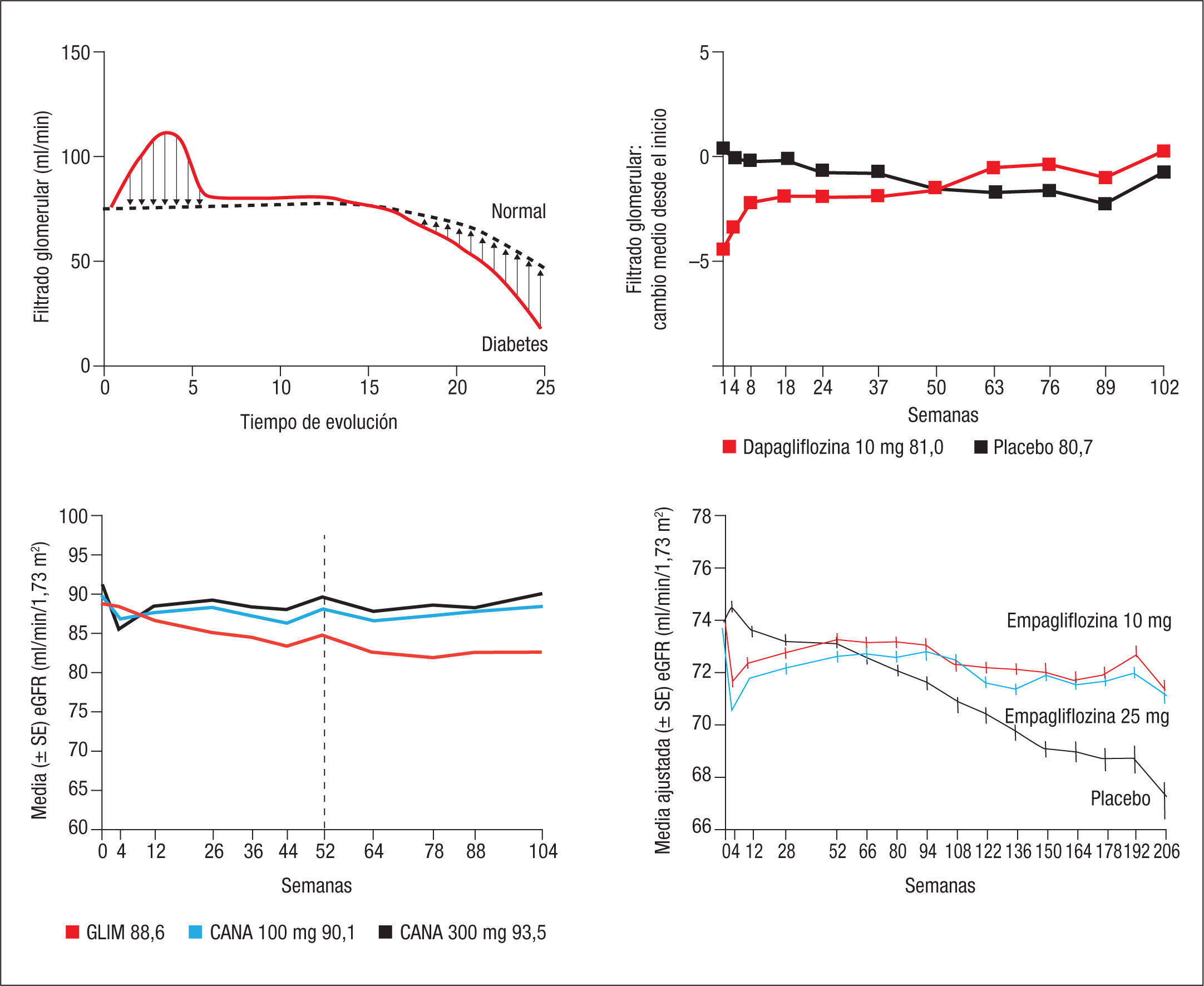
Los 3 iSGLT2 disponibles hasta ahora en Europa, dapagliflozina, empagliflozina y canagliflozina, tienen el mismo comportamiento sobre el filtrado glomerular: su inicio precoz se sigue de una normalización del filtrado previamente aumentado por la hiperfiltración32–34. A medio y largo plazo (2 años), el filtrado se mantiene en los pacientes tratados con iSGLT2 frente al de los pacientes tratados con placebo, cuya tendencia es ir perdiendo lentamente filtrado glomerular (fig. 7).

Efecto de los ÍSGLT2 sobre el filtrado glomerular: corrección inicial de la hiperfiitración detectada como un descenso transitorio del filtrado, seguido de normalización y finalmente mejoría sobre el filtrado glomerular de pacientes tratados con placebo de iSGLT2. Se observan los mismos resultados con dapagliflozina, canagliflozina y empaglifocina32–34. iSGLT2: inhibidores de SGLT2; SGLT2: cotransportador sodio-glucosa tipo 2.
El aumento de actividad de SGLT2 aumenta el transporte vectorial de glucosa y sodio, incrementando la reabsorción renal de glucosa.
Esta sobrecarga inesperada, pero constante, de glucosa supone una nueva fuente de glucotoxicidad, que no está sometida a oscilaciones prandiales ni a los ciclos diurnos-nocturnos, porque es constante, y es capaz de aumentar progresivamente a medida que aumenta la expresión de SGLT2 en el túbulo proximal.
Sin ninguna duda, este mecanismo, ignorado hasta hace pocos años, es una de las principales, si no la única, causas de la “pérdida de eficacia” observada con la mayoría de los antidiabéticos orales.
La inclusión de dapagliflozina en cualquier régimen terapéutico consigue hacer más duradero en el tiempo el efecto antiglucémico de dicho tratamiento, al devolver a una situación más próxima a la fisiológica la dinámica del manejo renal de la glucosa30,35.
Esta reducción en la sobrecarga diaria de glucosa obtenida con el uso de iSGLT2 tiene un efecto que no por lo evidente es menos sorprendente: reduce las necesidades de insulina, endógena o exógena, necesaria para el control metabólico. El resultado es una reducción en las demandas sobre la célula β y una menor resistencia periférica a la insulina. En los modelos experimentales de DM2, la utilización de dapagliflozina reduce la apoptosis de células p del islote de Langerhans, mejora su viabilidad y aumenta su capacidad de síntesis de insulina11 (fig. 3I y J). En pacientes con DM2 dependientes de insulina con altos requerimientos de esta, la utilización conjunta con dapagliflozina permite obtener las mismas dianas terapéuticas sin necesidad de incrementar paulatinamente las dosis de insulina36.
Señalizaciones alternativas de la insulina en la resistencia insulínica. Posible papel de los inhibidores de SGLT2A finales de los años noventa del pasado siglo, diversos estudios correlacionaron la resistencia a la insulina, medida como inhibición de la captación celular de glucosa, con la activación de vías metabólicas secundarias (hexosaminas) a partir del aumento de fructosa 6 fosfato37–39. Sin embargo, el concepto de resistencia a la insulina está experimentando profundos cambios conceptuales de la mano de estudios experimentales en los que se pueden bloquear de modo órgano-específico o tejido-específico los receptores de insulina, para estudiar los efectos de la ausencia de señalización de insulina a nivel de estos.
Es verdaderamente sorprendente ver cómo el “knock-out” de los receptores de la insulina en el tejido adiposo lleva a su atrofia40, de modo que el aumento de tejido adiposo ampliamente reportado en relación con la resistencia a la insulina en la DM2 no es atribuible a una reducción en la señalización a través de sus receptores en dicho tejido. “Resistencia a la insulina” no es equivalente a “menor sensibilidad de los receptores a la insulina”41.
La descripción del receptor de insulina queda más allá de los objetivos de este capítulo. Ha sido amplia y profusamente estudiado, y existen excelentes revisiones al respecto42,43. Baste con saber que la unión a su receptor activa la capacidad fosforilativa de este, que fosforila y activa un grupo de moléculas intermedias (los sustratos de los receptores de insulina 1RS1 e IRS2). 1RS1 e IRS2 inician una cascada de fosforilaciones y activaciones enzimáticas mediadas por la PKC-B y la fosfatidil-inositol 3 cinasa, cuyos resultados sobre la activación del transporte de glucosa (GLUT), de la síntesis y degradación de glucógeno y de ácidos grasos, de la glucólisis y gluconeogénesis son bien conocidos.
Sin embargo, la insulina, a través de esos mismos receptores, cumple en el organismo una función trófica, de crecimiento y supervivencia celular, que puede llegar incluso a entrar en conflicto con la señalización anterior.
La ocupación del receptor de insulina por esta puede activar vías proliferativas mediadas por G proteínas y activación de MAP-cinasa. Situaciones como la inflamación crónica, la producción de adipocitocinas por el tejido adiposo o el transporte exagerado de glucosa por la célula tubular renal activa cinasas como la ERK, JUNK o la PKC-C20, capaces, entre otras acciones, de fosforilar en posiciones equivocadas a IRS1 o IRS2. A través de esta segunda vía, el exceso de insulina puede reducir la señalización de la primera vía, cerrando un círculo en el cual una menor respuesta hipoglucemiante a la insulina provoca un aumento en la secreción de esta (o en la dosificación de insulina por el médico encargado), que a su vez empeora la resistencia al tiempo que aumenta los efectos proliferativos, tróficos y de supervivencia celular de la propia insulina.
A nivel hepático, estos efectos se traducen en un aumento en la síntesis de triglicéridos, ácidos grasos y ApoB y una reducción en la expresión de receptores a lipoproteínas de baja densidad41. A nivel del tejido adiposo, la insulina señalizando a través de esta vía no solo inhibe la lipólisis, sino que estimula la lipogénesis e hipertrofia del tejido graso, facilitando la infiltración inflamatoria de la grasa con generación de adipocitocinas41. A nivel endotelial, con condiciones que favorecen la resistencia a la insulina, la producción habitual de NO en respuesta a la insulina44 se sustituye por activación en la síntesis de endotelina41,45 (en el insulinoma, la producción de insulina es vasodilatadora). Aumenta la dotación de moléculas procarcinogénicas como UCAM-1 y VEGF, aumenta el depósito local de lípidos y disminuye el flujo sanguíneo hacia músculo y tejido adiposo46. A ni vel glomerular, la insulina es un factor de crecimiento y supervivencia: causa la desdiferenciación del podocito, la pérdida urinaria de nefrina y la fusión pedicelar25,41.
En definitiva, la activación de la vía de señalización de insulina de proliferación y supervivencia causa hipertrofia del tejido adiposo, aumento en el transporte renal de glucosa a través de SGLT2 con reducción del “feed-back” tubuloglomerular e hiperfiltración, activación de endotelina, catecolaminas y angiotensina II, junto con aumento en la reabsorción de sodio e hipertensión. También causa disminución en la disponibilidad de IRS activos, con la resistencia metabólica consiguiente.
¿Qué efectos cabe esperar de la inhibición de los SGLT2 más allá de la reducción en la sobrecarga calórica desde el riñón?La reducción en la génesis renal de ROS y AGE y la disminución en la señalización de la vía PKC reduce la fibrosis glomerular e intersticial, disminuye la activación inflamatoria, restablece el “feed-back” tubuloglomerular, y disminuye la vasodilatación aferente y la hiper-filtración.
La reducción en la resistencia a la insulina frena y revierte la esteatosis hepática, la hipertrofia del tejido adiposo, la producción de endotelina, la desdiferenciación del podocito y el agotamiento de la célula B.
Ambos efectos, probablemente, se combinan para explicar las importantes y precoces reducciones observadas en la mortalidad cardiovascular y la progresión del daño renal en la DM2 en el primer ensayo de seguridad cardiovascular publicado de uno de los fármacos de esta familia iSGLT-247.
Conflicto de interesesEl autor declara haber actuado como asesor de AstraZeneca para el desarrollo de dapagliflozina a través de los programas de formación REVERSE, Diabetter y Conexión; haber actuado como asesor de Otsuka para el desarrollo de tolvaptán; formar parte del European Hyponatremia Network para el desarrollo del conocimiento en hiponatremia; ser coordinador nacional del estudio DELIGHT de AstraZeneca, y haber licenciado una patente en nefroprotección a Spherium Biomed.
