





Editor: Francesc Cardellach
Editor asociado: Òscar Miró
Caso: 162-2008
La paciente tenía como antecedentes un neumotórax durante la juventud, una ooforectomía y salpingectomía por endometriosis, una apendicectomía; era portadora de una prótesis de rodilla izquierda y actualmente presentaba hipertensión arterial, hipercolesterolemia y una hernia de hiato, por las cuales realizaba tratamiento con enalapril, simvastatina y omeprazol. La paciente 2 años antes había iniciado sialorrea y leve disfagia a sólidos, por lo que fue estudiada en otro centro. En este estudio se encontró una masa en el mediastino posterior que dependía del esófago y un nódulo pulmonar en el lóbulo medio que se puncionó. Se diagnosticó de tumoración de origen incierto, con un previsible muy mal pronóstico a corto plazo, por lo que ella y la familia decidieron no continuar el estudio.
Dieciséis meses después, consultó en un segundo centro por progresión de la disfagia, que en aquel momento era moderada y permitía una dieta de productos líquidos, semilíquidos y triturados. El estado general estaba conservado. Se realizó una fibrogastroscopia en la que se observó estenosis esofágica media y distal por una masa con mucosa de aspecto conservado que se extendía desde el esófago medio hasta el estómago proximal. En la biopsia se observó acumulación de fibrina y granulocitos neutrófilos, material necrótico, microfragmentos de epitelio escamoso esofágico sin atipias y cambios propios de esofagitis por reflujo. Se realizó una tomografía computarizada (TC) toracoabdominal, en la que se observó una masa mediastínica posterior que dependía del esófago, era excéntrica y causaba irregularidad en la luz esofágica; había también derrame pleural bilateral, una imagen nodular en el campo medio del pulmón derecho y cicatrices en el ápex. Se observaron adenopatías en la zona de la curvatura menor de la unión esofagogástrica y quistes renales. Se realizó una fibrobroncoscopia que mostró traqueobroncomalacia y una mucosa bronquial difusamente inflamada, pero sin lesiones indicativas de neoplasia. La citología del lavado broncoalveolar fue negativa para células tumorales y sus cultivos (en medios aerobio y anaerobio, para hongos y de Lowënstein) también fueron negativos. Una ecoendoscopia digestiva mostró una masa heterogénea, apolillada y estenosante, y en el fórnix gástrico se observó una masa protruyente en la zona cardial que se biopsió, aunque tampoco resultó diagnóstica. La paciente decidió no proseguir el estudio.
Ocho meses después, la paciente consultó en el servicio de medicina interna del Hospital Clínic al agravarse la disfagia a sólidos y añadirse disfagia a líquidos. Desde hacía una semana presentaba vómitos posprandiales inmediatos, anorexia y en los últimos 2 meses había perdido unos 10 kg de peso. La paciente ingresó para estudio. En la exploración física destacaba edema blando en las extremidades inferiores. En la radiografía posteroanterior de tórax se observaba cardiomegalia. En el electrocardiograma había un ritmo auricular caótico. En la analítica de sangre se obtuvieron los resultados siguientes: velocidad de sedimentación globular 103 mm/h, hematíes 3,28 3 1012/l, hemoglobina 100 g/l, hematocrito 0,31 l/l, volumen corpuscular medio 95 fl, hemoglobina corpuscular media 30 pg, leucocitos 15,7 3 109/l (78% segmentados, 2% no segmentados, 1% eosinófilos, 15% linfocitos, 4% monocitos), plaquetas 220 3 109/l, actividad de protrombina 77%, glucemia 148 mg/dl (8,2 mmol/l), nitrógeno ureico en sangre 11 mg/dl (1,7 mmol/l), creatinina 0,8 mg/dl (70 mmol/l), sodio 135 mmol/l, potasio 4,6 mmol/l, calcio 8,5 mg/dl (2,1 mmol/l), fósforo 3,4 mg/dl (1,1 mmol/l), sideremia 18 mg/dl (3,2 mmol/l), aspartato aminotransferasa 12 U/l, alanino aminotransferasa 15 U/l, lactato deshidrogenasa 381 U/l (n: 250-450), gammaglutamiltranspeptidasa 106 U/l, fosfatasa alcalina 533 U/l, bilirrubina total 0,4 mg/dl (6,8 mmol/l), amilasa 39 U/l, lipasa 22 U/l, colesterol 126 mg/dl (1,5 mmol/l), triglicéridos 92 mg/dl (1,0 mmol/l), creatincinasa 25 U/l, proteínas totales 56 g/l (albúmina 52%, alfa1 5%, alfa2 12%, beta 15%, gammaglobulina 16%). Se realizó una TC toracoabdominal en la que se observó una masa extensa que afectaba a los tercios medio y distal del esófago hasta alcanzar el cardias, con áreas de necrosis en su interior, que condicionaba una dilatación de la luz proximal. Presentaba una imagen de calcificación, aunque podría ser también contraste oral de una prueba previa. Había varios nódulos de pequeño tamaño en ambos pulmones, el de mayor dimensión localizado en el lóbulo medio con imagen de broncograma aéreo, y discreto derrame pleural bilateral. En el abdomen se identificó una adenopatía retrogástrica de 2 cm de diámetro y otras de ta-maño inferior a 1 cm, adyacentes al cuerpo pancreático, retrocrurales, paraaórticas y mesentéricas.
A continuación se realizó una exploración que resultó diagnóstica.
Dra. Maica Galán. En resumen, se trata de un mujer de 78 años con disfagia progresiva de lenta evolución, con estado general conservado hasta 2 meses antes de su ingreso, y con una masa en el mediastino posterior que parece depender de esófago. Antes de proceder a la discusión del caso, agradecería los comentarios del radiólogo.
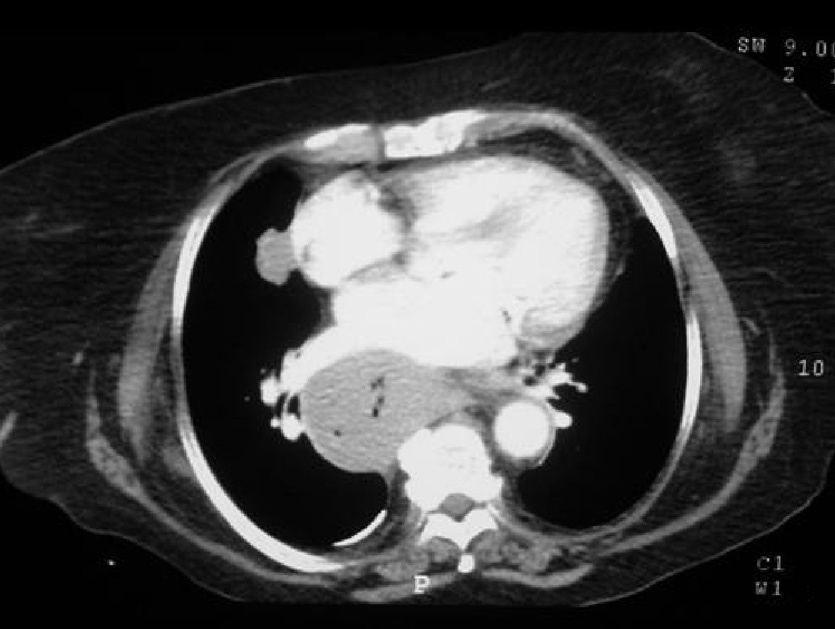
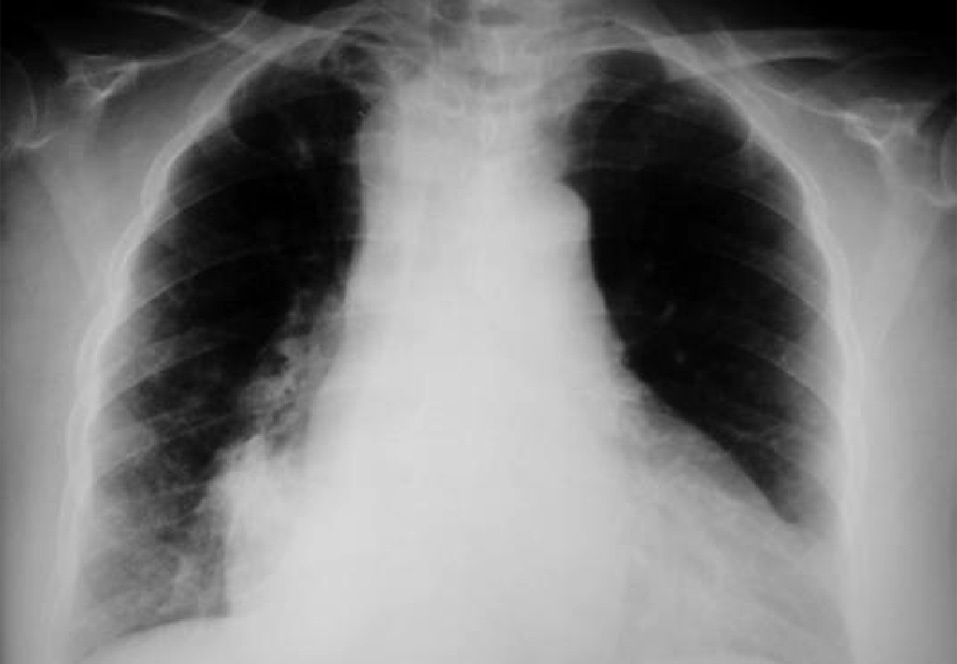
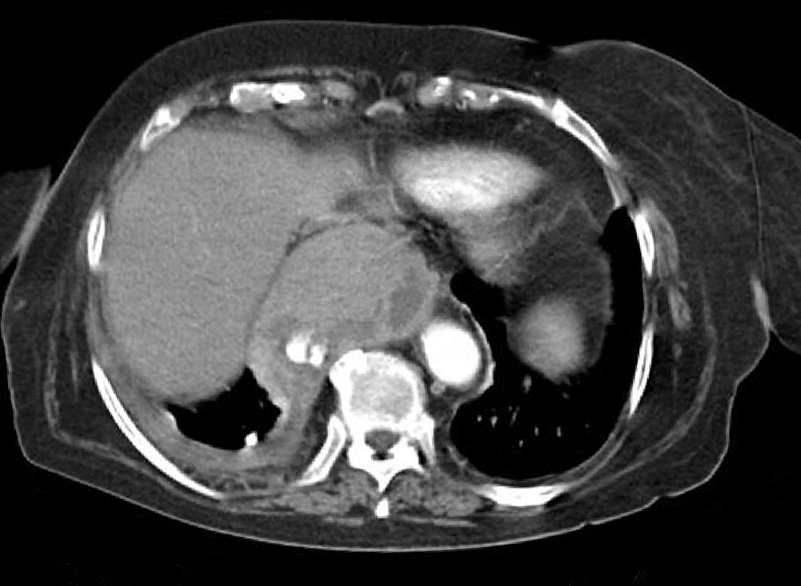
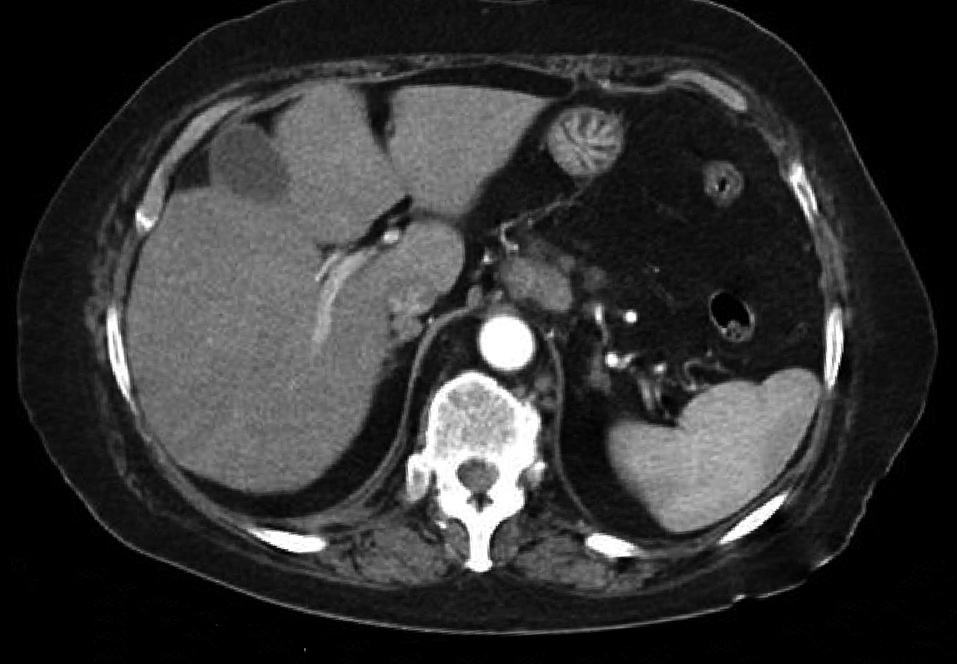
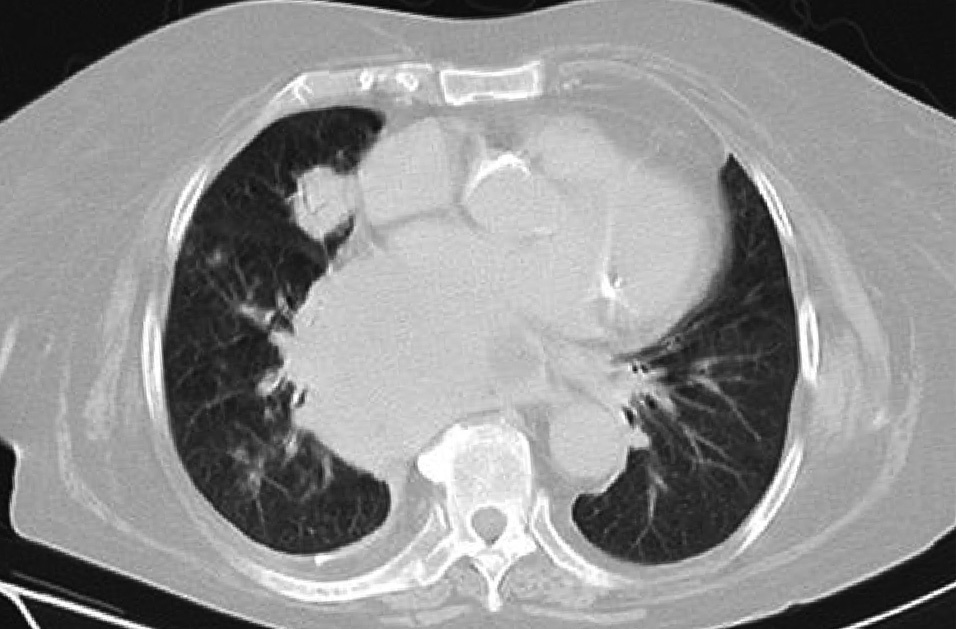
Dr. Màrius Pagés. La TC toracoabdominal realizada en el segundo centro mostró una masa mediastínica posterior que dependía del esófago y que afectaba al tercio medio y distal (fig. 1). Se observaba también un nódulo pulmonar en el lóbulo medio derecho y algunas cicatrices en el ápex. Se detectaron también adenopatías en la zona de la curvatura menor de la unión esófago-gástrica. En la radiografía posteroanterior de tórax se observaba cardiomegalia (fig. 2). La TC toracoabdominal realizada en nuestro centro mostró la masa extensa en los tercios medio e inferior esofágicos hasta alcanzar el cardias, que mostraba áreas de necrosis en su interior (fig. 3) y adenopatías en la región celíaca (fig. 4). Se observaron varios nódulos de pequeño tamaño en ambos pulmones y uno de mayores dimensiones en el lóbulo medio con imagen de broncograma aéreo (fig. 5).
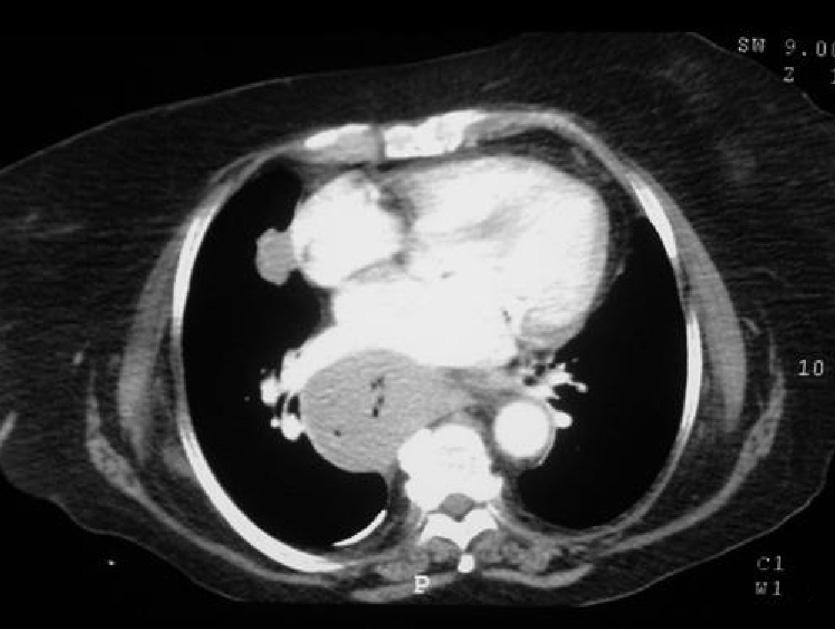
Fig. 1. Tomografía computarizada (TC) de tórax que muestra una masa sólida en el tercio medio esofágico. Se observa, además, un nódulo pulmonar enlóbulo medio.
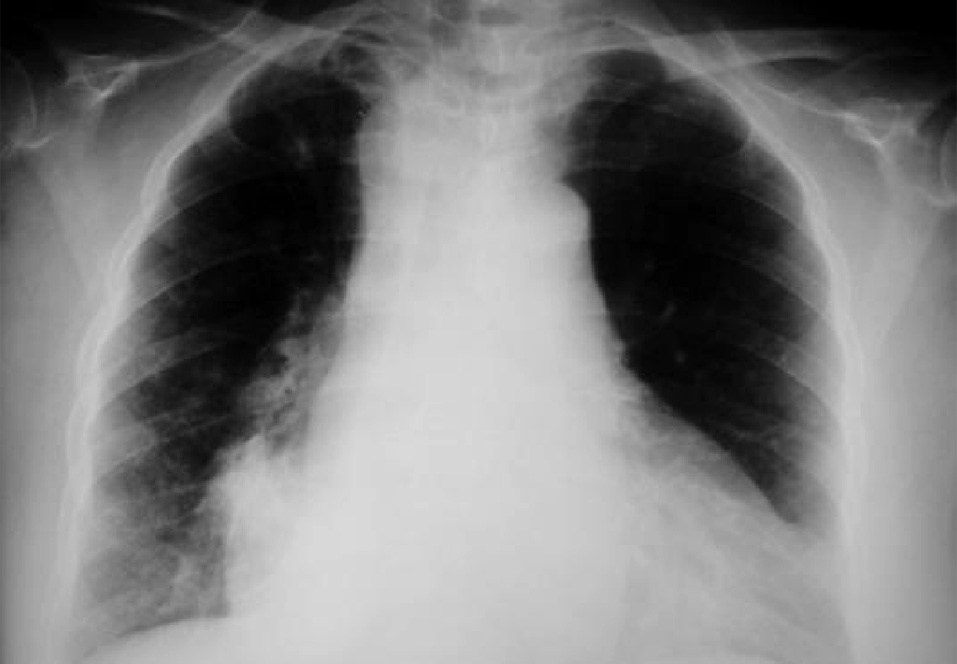
Fig. 2. Radiografía de tórax que muestra cardiomegalia.
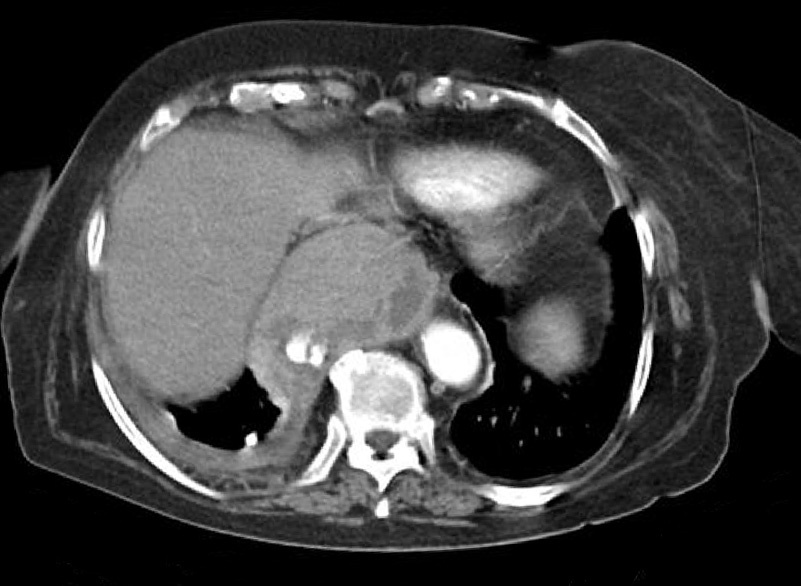
Fig. 3. Tomografía computarizada (TC) de tórax. La masa esofágica se extiende hacia el cardias y se observan áreas de necrosis.
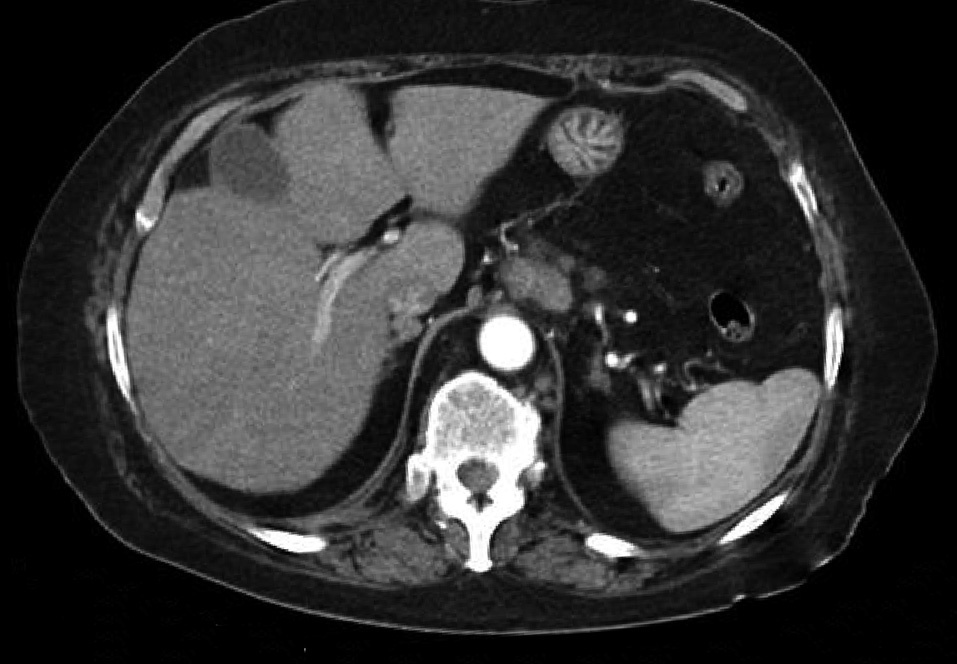
Fig. 4. Tomografía computarizada (TC) abdominal que muestra una adenopatía de tamaño significativo en la región celíaca.
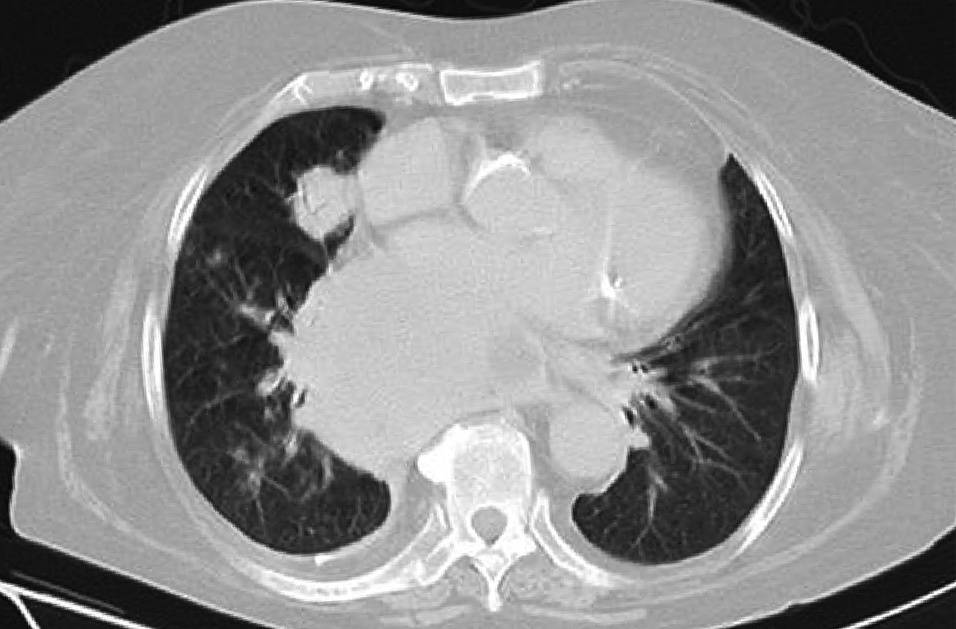
Fig. 5. La tomografía computarizada (TC) de tórax con ventana de pulmón detecta broncograma aéreo en el nódulo pulmonar del lóbulo medio, así como otros nódulos adicionales de tamaño menor.
Dra. M. Galán: Se trata de una mujer de 78 años que, en el momento del ingreso, presenta 2 cuadros clínicos. El primero basado en el síntoma principal de la paciente, la disfagia, que destaca por su progresiva, pero lenta evolución. El segundo, por la presencia de edemas en las extremidades inferiores, cardiomegalia y ritmo caótico auricular, un cuadro de insuficiencia cardíaca.
La iconografía muestra una masa en mediastino posterior que depende de esófago, concretamente de la submucosa, muy homogénea en su porción más proximal, pero con áreas de necrosis compatible con degeneración maligna en la distal. Por otro lado, destaca un nódulo pulmonar de tamaño considerable en lóbulo medio, así como nódulos más pequeños en la última TC. No hay calcio ni grasa en los nódulos pulmonares como para pensar en una enfermedad granulomatosa o en un hamartoma y, por tanto, debemos considerar el diagnóstico de metástasis pulmonares. Se aprecia además una adenopatía retrogástrica de 2 cm y otras inespecíficas. En la fibrogastroscopia y la ecoendoscopia digestiva realizadas en el segundo centro, se describe una masa estenosante y apolillada, con mucosa conservada, que se extiende desde el esófago medio al estómago proximal, con 2 biopsias negativas para tumor. La descripción como masa apolillada iría a favor de necrosis y, por lo tanto, de degeneración maligna.
Me gustaría desarrollar un diagnóstico diferencial preciso, pero sencillo, y para ello me voy a centrar en el síntoma principal de la paciente, que es la disfagia. Cuando un paciente presenta dificultad para la deglución, debemos pensar que la causa puede ser orofaríngea o esofágica1.
En el caso de esta paciente, y dada la ayuda de la imagen radiológica y endoscópica, nos debemos centrar en la segunda. La disfagia de causa esofágica puede ser de 2 tipos1: orgánica o mecánica, debida a estenosis de la luz esofágica, que se caracteriza por ser predominantemente a sólidos, continua y progresiva, y funcional o motriz, por alteración de la peristalsis esofágica y/o incoordinación en la relajación del esfínter esofágico inferior, que es predominantemente a líquidos e intermitente.
Dado que se aprecia una masa en mediastino posterior por TC y teniendo en cuenta las características de la disfagia, creo que la causa es mecánica y no motriz.
Entre las causas de disfagia mecánica, cabe destacar1: a) las intraluminales (cuerpos extraños y membranas/anillos esofágicos); b) las intramurales (inflamación aguda, infecciosa, cáusticos, reflujo gastroesofágico, amiloidosis y neoplasias benignas o malignas de esófago), y c) la compresión extrínseca por masas mediastínicas (bocio, timoma, linfoma, sarcoidosis), crecimiento auricular izquierdo, vascular como la disfagia aórtica o la disfagia lusoria, neoplasias broncopulmonares, principalmente izquierdas, y divertículos esofágicos grandes.
La imagen de la TC y la ecoendoscopia descartan la etiología intraluminal y, razonablemente, la compresión extrínseca. Por ello, me voy a centrar en la disfagia esofágica de causa mecánica e intramural, con masa en el mediastino posterior que depende de esófago y, concretamente, en las neoplasias benignas y malignas de esófago.
La Organización Mundial de la Salud (OMS) clasifica los tumores del esófago en2,3:
1. Tumores que secundariamente afectan al esófago o metástasis. Son muy extrañas, la mayoría se localiza en el tercio medio esofágico, suelen ser masas submucosas sin invasión de la mucosa y el síntoma más frecuente es la disfagia. Se han descrito metástasis de tumores primarios como tiroides, pulmón, mama, riñón, próstata y ovario, y son, por tanto, indicadores de diseminación sistémica. Otro ejemplo sería la metástasis de melanoma en el esófago que, aunque infrecuente, es más común que el primario de esófago. Yo no profundizaría en el tema, dada la escasa frecuencia y el comportamiento clínico tan agresivo, a diferencia del tumor que presenta nuestra paciente.
2. Los tumores primarios de esófago los podemos clasificar en dos grandes grupos: epiteliales y no epiteliales. Voy a empezar revisando los de tipo epitelial, que son los más frecuentes en la práctica clínica habitual, dado que incluyen el carcinoma escamoso y el adenocarcinoma4. Es importante recordar que desde los años 1980, tanto en EE.UU. como en Europa occidental, la incidencia de la variedad escamosa se mantiene estable, mientras que la del adenocarcinoma de esófago y del cardias ha aumentado sustancialmente. El carcinoma escamoso es la histología que predomina en el esófago cervical, así como en el esófago torácico superior y medio. Es más frecuente en varones y suele estar relacionado con hábitos tóxicos (tabaco y alcohol). El adenocarcinoma predomina en el esófago distal, también es más frecuente en varones, pero se relaciona con el reflujo gastroesofágico crónico y el esófago de Barrett.
La propagación tumoral de ambas entidades se hace principalmente por contigüidad y vía linfática. La diseminación linfática se demuestra en más del 70% de los pacientes con cáncer epitelial de esófago, y produce metástasis en ganglios mediastínicos, abdominales y cervicales. La diseminación hemática es más tardía.
La paciente tiene antecedentes de hernia de hiato en tratamiento con omeprazol, y en la biopsia realizada por fibrogastroscopia se aprecian cambios propios de esofagitis por reflujo. Si pensáramos en un tumor de estirpe epitelial, dado el antecedente de reflujo gastroesofágico, habría que pensar en un adenocarcinoma sobre un esófago de Barrett. De todas formas, hay muchos datos en contra de esta histología, como son el sexo, la edad, la lenta evolución clínica, una mucosa indemne en la exploración por endoscopia digestiva y la imagen radiológica. Igualmente, la histología de carcinoma escamoso la descartaría por todos estos motivos, a lo que se sumaría la ausencia de hábitos tóxicos.
Se han descrito otras variedades de estirpe epitelial incluidas dentro de los carcinomas2,3, como son el carcinoma poco diferenciado, el carcinoma de célula pequeña, el carcinoma escamoso con rasgos sarcomatoides, el carcinoma adenoide quístico, el carcinoma mucoepidermoide, el carcinoma verrugoso y el carcinoma de células basalioides. No me voy a entretener en ellos, pues se trata de tumores muy poco frecuentes, con afectación de la mucosa y comportamiento agresivo.
Dejando de lado a los carcinomas, hay otro subgrupo dentro de los tumores de estirpe epitelial2,3. Se trata de los tumores endocrinos, que son raros en el esófago, y, entre ellos, el carcinoma neuroendocrino de célula pequeña (pobremente diferenciado) y el tumor carcinoide (bien diferenciado). Del carcinoma neuroendocrino de célula pequeña se consideran frecuencias entre el 0,05 y el 7,6% de los cánceres esofágicos, el doble de frecuente en varones, aparece en la sexta década de la vida, generalmente en relación con el hábito tabáquico y también con el esófago de Barrett. Normalmente, se localizan en el tercio inferior esofágico, frecuentemente se diagnostican en estadios avanzados, se suelen presentar como una masa ulcerada y de crecimiento fúngico, y se asocian a secreción ectópica de sustancias (secreción inadecuada de hormona antidiurética [SIADH], hipercalcemia, diarreas, síndrome de hipopotasemia-aclorhidria, secreción de péptido intestinal vasoactivo [VIP]). El pronóstico es pobre, normalmente la supervivencia es inferior a 6 meses.
El segundo grupo es el de los tumores carcinoides5,6. En una revisión de 13.715 tumores carcinoides llevada a cabo por el registro norteamericano SEER6, que abarca de 1950 a 1999, el 67,5% de los casos se localizaba en el tracto gastrointestinal, y dentro de éstos, la mayoría se originaba en el intestino delgado (41,8%), seguido del recto (27,4%) y el estómago (8,7%). De todos los tumores carcinoides revisados, sólo 6, que corresponde al 0,06%, se originaban en el esófago. Eso representa el 0,05% del total de carcinoides del tracto gastrointestinal y el 0,02% de los cánceres de esófago. En el global de tumores carcinoides, hay un ligero predominio en las mujeres (55,5%), y esta diferencia es mayor en algunas localizaciones, como apéndice (68%) o estómago (63%), aunque se demostró un predominio evidente en varones en los casos que afectaban el esófago (66%) y el timo (76%). Los tumores carcinoides son más frecuentes entre los 56 y los 67 años. Está descrito un caso de un paciente con tumor carcinoide asociado a adenocarcinoma y esófago de Barret. Se suelen localizar en el tercio inferior del esófago y tienen un crecimiento lento hasta convertirse en grandes masas, que infiltran en profundidad la pared del esófago.
Es difícil descartar el tumor carcinoide de entrada porque, a pesar de que la frecuencia de aparición en el esófago es extraordinariamente baja y que normalmente aparecen en pacientes más jóvenes y producen ulceración de la mucosa, es cierto que están descritos casos de tumores submucosos y, por otro lado, suelen tener una evolución lenta que hace que puedan alcanzar un tamaño considerable. No puedo olvidar también que sería un tipo de tumor que ofrecería la posibilidad de justificar una relación entre los 2 cuadros clínicos que presenta la paciente (disfagia e insuficiencia cardíaca), por la asociación que hay entre tumor carcinoide y cardiopatía derecha7-9. En contra de ello, la paciente no presenta síndrome carcinoide, que suele estar presente en los casos de afectación cardíaca. El síndrome carcinoide, que ocurre en menos del 10% de los pacientes con tumores carcinoides, típicamente del intestino medio, y salvo algunas excepciones, sólo se manifiesta en presencia de metástasis hepáticas o, en ocasiones, pulmonares. Las manifestaciones cardíacas ocurren en el 33% de los pacientes y consisten en una fibrosis endocárdica, principalmente en las cavidades derechas, que provoca estenosis en la válvula pulmonar e insuficiencia tricuspídea, que suele desembocar en fallo cardíaco. Nuestra paciente presenta cuadro de insuficiencia cardíaca, clinicorradiológico, pero no presenta soplo cardíaco que nos haga sospechar insuficiencia tricuspídea.
Si nos olvidamos por un momento de los tumores de esófago de estirpe epitelial, nos hemos de centrar en los tumores mesenquimales y, aunque menos frecuentes, en los de origen hematológico. Voy a empezar describiendo los hematológicos y dejaré los mesenquimales para el final.
Se trata de linfomas no hodgkinianos (LNH)2,3,10-12. El tubo digestivo es el lugar de afectación extraganglionar más frecuente de los LNH. Predominan en el estómago (50-60%), y el esófago es la localización del tracto digestivo donde más infrecuentemente se asienta un linfoma (< 1% de los pacientes afectados de linfoma). Es más frecuente en varones alrededor de los 50 años y predominan en el tercio inferior del esófago. Los que se originan en el estómago suelen cursar con epigastralgia, anorexia, sensación de saciedad exagerada e inmediata tras la ingesta y, eventualmente, hemorragia digestiva. Se asientan preferentemente en el antro, seguido del cuerpo y el cardias. Suelen ser linfomas de célula grande tipo B o linfomas MALT (tejido linfoide asociado a mucosas) de célula B de bajo grado. Los primeros suelen tener una evolución rápida, a diferencia de los MALT, que son de evolución más lenta. A diferencia de lo que le ocurre a nuestra paciente, su diagnóstico por biopsia endoscópica no suele ser complicado.
El tumor mesenquimal2,3 más frecuente de esófago es el leiomioma, que a su vez es también, con gran diferencia, el más frecuente entre los tumores benignos de esta localización. Son el doble de frecuentes en varones y la mediana de edad es de 30-35 años. Los sarcomas de esófago representan el 0,2% de los tumores esofágicos malignos, los varones resultan el doble de afectados y acontece entre la sexta y octava décadas de la vida. Los tumores del estroma esofágico tienen características demográficas similares a los sarcomas.
El leiomioma4 es un tumor submucoso que crece a partir de la capa muscular circular del esófago y, en el 80% de los casos, se localiza en el esófago distal. Suelen llegar a ser tumores de gran tamaño que se extienden al mediastino, con un patrón de crecimiento lento. Habitualmente no provocan síntomas, aunque a veces se manifiestan con disfagia y dolor torácico, sobre todo los tumores con un tamaño superior a los 5 cm. El diagnóstico se realiza mediante examen radiológico y endoscópico.
Otros tumores benignos de esófago2,3 son los lipomas y los tumores de células granulares. Los primeros suelen aparecer en el tercio superior esofágico y son de fácil diagnóstico por la imagen, al observarse grasa en su interior. Los tumores de células granulares suelen presentarse como nódulos o pólipos sesiles pequeños, predominantemente en el esófago inferior. Está descrito algún caso de degeneración maligna. También hay una entidad, la leiomiomatosis múltiple, que se presenta más frecuentemente en adolescentes y adultos jóvenes, a veces con extensión distal a la porción superior del estómago. Puede ser familiar, asociada a la nefropatía de Alport, y también se han descrito otras asociaciones, como leiomiomas uterinos o vulvares, así como leiomiomatosis traqueobronquial.
La mayoría de los sarcomas2,3 se proyectan dentro de la luz. El patrón endoscópico es de un tumor submucoso con protusión en una mucosa normal. Los sarcomas, la mayoría representados por los tumores del estroma gastrointestinal (GIST), son típicamente multinodulares o, menos frecuentemente, se presentan como masas en placas, semejante a los sarcomas de partes blandas.
La variedad maligna del leiomioma es el leiomiosarcoma2-4,13, con varios grados de diferenciación en función del número de mitosis. El leiomiosarcoma resulta ser menos del 1% de las neoplasias malignas de esófago y alrededor del 4% del global de tumores derivados de músculo liso y estroma. Aparece en adultos añosos, normalmente se localiza en el esófago inferior y típicamente se presenta como una gran masa con hemorragia y necrosis en su interior. Puede derivar en metástasis, principalmente pulmonares y hepáticas, y, aunque raramente, también en adenopatías. La sintomatología suele ser similar a la de otras neoplasias esofágicas, aunque la clínica de presentación es mucho más larvada, debido al lento crecimiento de estos tumores. La biopsia endoscópica14 conlleva un elevado número de falsos negativos, principalmente si la mucosa esofágica está intacta.
Otra entidad englobada dentro de los tumores submucosos y de origen mesenquimal sería el GIST2,3,15-19. Representa menos del 3% de las neoplasias gastrointestinales, pero es el tumor mesenquimal más frecuente del tracto digestivo. La mayoría de GIST se origina en el estómago (50-60%) y en el intestino delgado (30%), mientras que los tumores esofágicos y de colon-recto son relativamente poco frecuentes (aproximadamente un 5% cada uno). Los GIST también se pueden originar de localizaciones abdominopélvicas extraintestinales, como son el omento, el mesenterio y el retroperitoneo. Pueden coexistir varios GIST. Suele aparecer en adultos entre la sexta y octava décadas de la vida. Aunque muy raramente, están descritos casos en niños y adultos jóvenes, que suelen formar parte de la tríada de Carney (GIST o leiomiosarcoma de la antigua clasificación, paraganglioma y condromas pulmonares). Hasta el año 2000, el número de casos de GIST había estado subestimado y poco publicado. Actualmente, el diagnóstico preciso de GIST es muy importante para el tratamiento correcto del paciente. Se trata de tumores morfológicamente heterogéneos, con un subgrupo de células fusiformes que es el más frecuente (70% de los casos) y que es el grupo de tumores que antes del año 2000 se clasificaban como leiomiosarcomas. Hay otro subgrupo de células epitelioides (el restante 30%) y es el que previamente se diagnosticaba como leiomioblastoma, a pesar de que también con frecuencia se podía confundir con un carcinoma poco diferenciado, tumor carcinoide o linfoma. El GIST se define por la expresión de un receptor del factor de crecimiento de tirosina cinasa, CD117, también conocido como cKIT. Aproximadamente, el 20% de los casos tiene una presentación metastásica que afecta con más frecuencia al hígado o peritoneo. La diseminación linfática es muy rara. La mayoría de lesiones nodales son por depósitos metastásicos en el omento o peritoneo, más que verdadera diseminación linfática de la enfermedad. El riesgo de los pacientes se evalúa de acuerdo al esquema de consenso publicado en 2002, basado en el tamaño del tumor y el recuento de mitosis. También se han realizado avances importantes en las evaluaciones moleculares, ya que sabemos que el lugar de la mutación de cKIT se correlaciona con la respuesta a imatinib.
Dentro de los tumores mesenquimales no hay que olvidar los tumores neurogénicos20,21. Son las neoplasias primarias más frecuentes en el mediastino posterior, de las cuales el 6-28% son malignas y se agrupan dentro de los sarcomas de partes blandas. Cuando su origen está en la célula de cubierta de los nervios periféricos (célula de Schwann), se denominan schwannomas, con una variante benigna y otra maligna, y a menudo se asocian a la neurofibromatosis de Von Recklinghausen, aunque está descrito algún caso esporádico.
Otros sarcomas son mucho menos frecuentes2,3. Los rabdomiosarcomas son típicos en la infancia, aunque se ha descrito algún caso en adultos. Se originan en el músculo estriado y predominan en el esófago distal. El sarcoma sinovial, descrito en niños y adultos añosos, suele presentarse como una masa polipoide en esófago proximal. Los pacientes con síndrome de inmunodeficiencia adquirida pueden presentar sarcoma de Kaposi que afecte al esófago. Otras entidades excepcionales en el esófago son el tumor miofibroblástico inflamatorio y el tumor desmoide.
Finalmente, y a modo de resumen, he desarrollado el diagnóstico diferencial de las causas tumorales que condicionan la disfagia esofágica mecánica, y me he basado en la existencia de una masa en mediastino posterior que depende del esófago. Por la descripción de la TC toracoabdominal, asumo que nuestra paciente tiene metástasis pulmonares y alguna adenopatía locorregional. Por ello, descarto los tumores benignos, como serían el leiomioma, el lipoma o el schwannoma. También por la descripción de lagen submucoso, por lo que quedarían descartados el adenocarcinoma y el carcinoma escamoso de esófago, así como todas las variantes epiteliales anteriormente mencionadas. Teniendo en cuenta que nuestra paciente tiene disfagia de 2 años de evolución y un estado general conservado, a pesar del gran tamaño y extensión del tumor, descarto los tumores de comportamiento muy agresivo, como podrían ser las metástasis, el carcinoma neuroendocrino de célula pequeña, así como algunos sarcomas. No puedo descartar el tumor carcinoide, porque se ajusta al comportamiento clínico, la evolución lenta y la imagen radiológica, por no decir que sería el único que de forma lógica conseguiría relacionar los 2 cuadros clínicos descritos en nuestra paciente. Pero no debemos olvidar que se ha descrito de forma excepcional en el esófago, e iría en contra del diagnóstico la ausencia de diseminación hepática, de síndrome carcinoide y de afectación mucosa esofágica.
Creo que la paciente está afectada de un tumor esofágico de origen submucoso y no epitelial, con características de degeneración maligna. Podría tratarse de un tumor mesenquimal de tipo leiomiosarcoma (por degeneración de un leiomioma) o un tumor del estroma gastrointestinal tipo GIST, ambos difíciles de diferenciar desde el punto de vista clínico, radiológico y endoscópico, y, hasta hace pocos años, solapados también desde el punto de vista histológico.
Para concluir, considero que la exploración que se realizó para alcanzar el diagnóstico fue repetir una endoscopia digestiva alta con biopsia.
Diagnóstico de la Dra. M. Galán
Neoplasia esofágica no epitelial, probablemente mesenquimal (leiomiosarcoma o GIST).
DIAGNÓSTICO CLÍNICO
Tumor esofágico no epitelial.
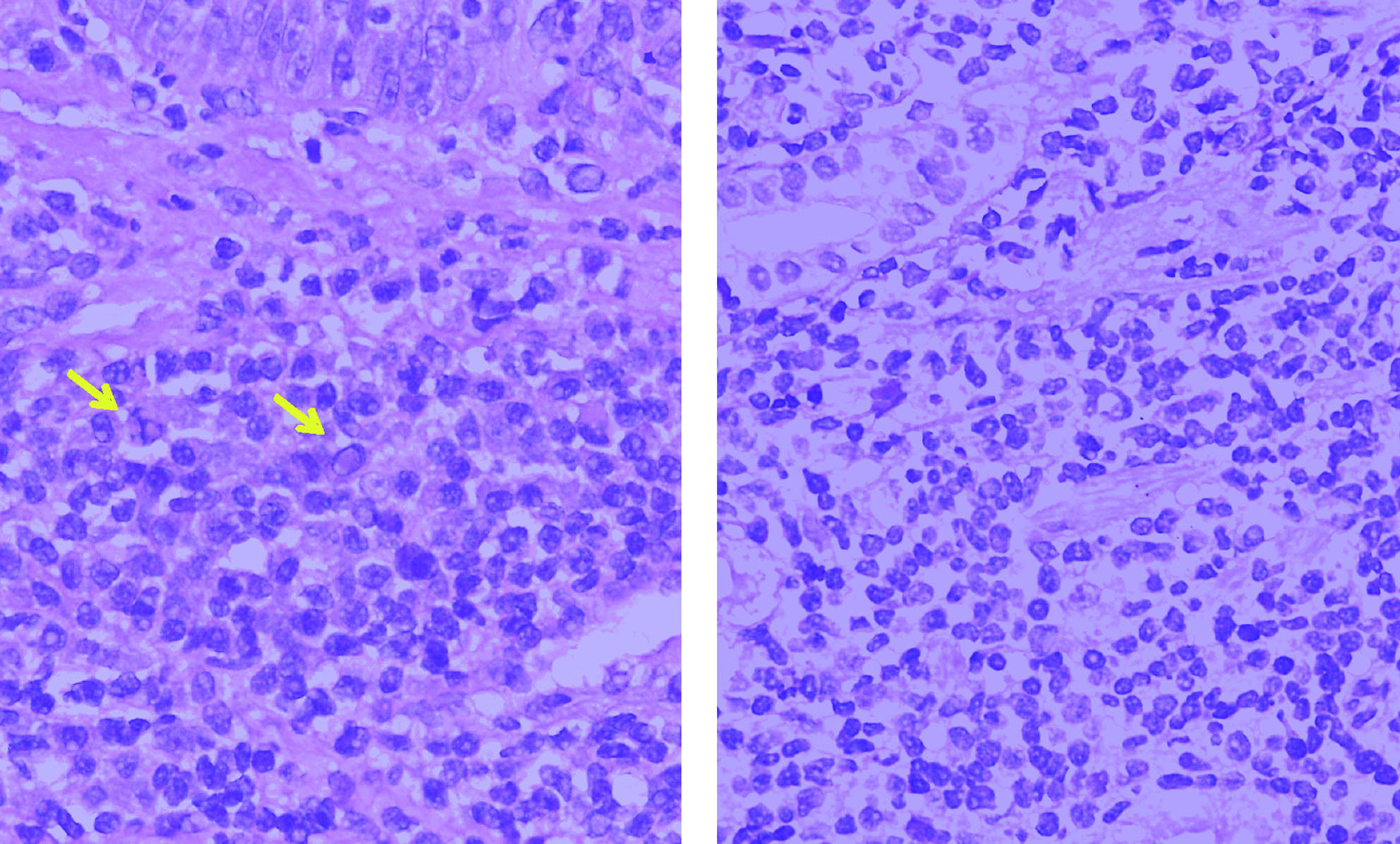
Dr. Lluís Colomo. Las biopsias remitidas y obtenidas durante una nueva exploración al Servicio de Anatomía Patológica correspondían a 3 pequeños fragmentos de mucosa esofágica y a un fragmento de mucosa glandular. En todos ellos se reconocía la presencia de un infiltrado linfoide localizado en lámina propia. Citológicamente, estaba constituido por una población de linfocitos pequeños, con clara diferenciación plasmocelular en las biopsias esofágicas y algunas células con seudoinclusiones intranucleares (cuerpos de Dutcher) correspondientes a agregados de inmunoglobulinas (Ig). El fragmento con mucosa glandular no mostraba lesiones linfoepiteliales ni estructuras baciliformes con morfología compatible con Helicobacter pylori (fig. 6).
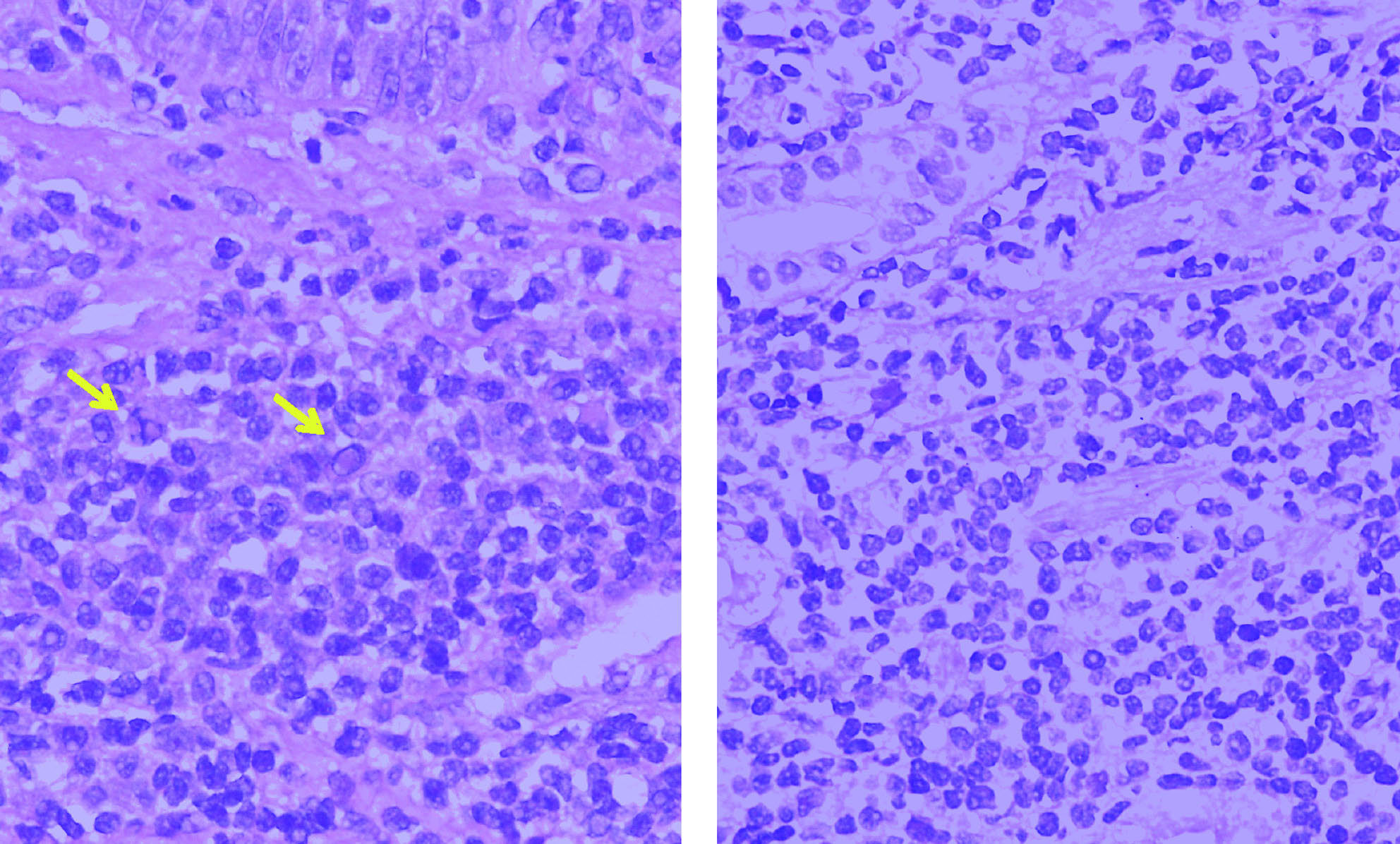
Fig. 6. Infiltrado linfoide con diferenciación plasmocelular y ocasionales cuerpos de Dutcher (flechas) (HE3 400).
El estudio inmunohistoquímico demostraba que la población linfoide expresaba marcadores de línea B (CD20) y presentaba restricción de cadenas ligeras kappa, con expresión de la cadena pesada de las IgM (fig. 7). No se expresaban otras cadenas pesadas, así como tampoco otros marcadores (CD5, CD43, CD56 o CD3). El índice proliferativo de la neoplasia, valorado con el anticuerpo Ki67, era bajo (10-20%).

Fig. 7. Infiltrado neoplásico que expresa CD20 y muestra la restricción de cadena ligera kappa, con expresión de cadena pesada de la inmunoglobulina M.
Todos estos rasgos morfológicos y fenotípicos, junto a la información clínica disponible en ese momento, indicaron el diagnóstico de linfoma B de bajo grado con diferenciación plasmocelular, y se planteó un diagnóstico diferencial entre un linfoma de la zona marginal extranodal (linfoma MALT) y un linfoma linfoplasmacítico. Esta situación de diagnóstico diferencial entre las 2 entidades no es infrecuente, dado que ambas presentan rasgos morfológicos y fenotípicos superpuestos, por lo que la información clínica, las características patológicas y las alteraciones genéticas deben integrarse para intentar diferenciar estos tumores. El linfoma de la zona marginal se presenta usualmente en territorios extraganglionares, aunque puede haber afectación ganglionar secundaria y existen también formas ganglionares primarias. La forma de presentación más frecuente es la gástrica, en el contexto de gastritis crónica relacionada con H. pylori, con menor incidencia de afectación de otros territorios (pulmonar 14%; área otorrinolaringológica 14%; ojo y tejido periorbitario 12%; piel 11%; tiroides 4%)22. En un 25% de las formas primarias gástricas, y en hasta un 46% de formas extragástricas, se ha descrito enfermedad con afectación multiorgánica en el momento del diagnóstico23. La afectación de médula ósea varía entre un 2 y un 28% en las series publicadas23,24 y parece relacionarse con localizaciones extragástricas. El linfoma linfoplasmacítico es una entidad no demasiado bien caracterizada en la clasificación de la OMS, debido a que hasta el momento no se conocen alteraciones genéticas o rasgos fenotípicos específicos y a su solapamiento conceptual con la macroglobulinemia de Waldeström25. Los criterios actualmente propuestos para establecer el diagnóstico de macroglobulinemia de Waldeström son los siguientes: gammapatía monoclonal IgM a cualquier concentración, infiltración intersticial o nodular por linfocitos pequeños, células plasmacitoides y células plasmáticas, y un inmunofenotipo CD19+ CD20+ CD5- CD10- CD23-, con presencia de Ig de superficie26. En las recientes reuniones de actualización de la clasificación de la OMS, se ha acordado considerar macroglobulinemia de Waldeström como un subgrupo de linfoma linfoplasmacítico, considerando esta última entidad como un linfoma con los rasgos morfológicos y fenotípicos descritos, con o sin gammapatía monoclonal, y con la condición de excluir otros tipos de linfomas (Dr. E. Campo, comunicación personal). La ausencia de alteraciones genéticas y fenotípicas específicas seguirá dificultando el diagnóstico de esta entidad27.
Volviendo a esta paciente, las exploraciones realizadas después de la biopsia mostraron que no había afectación de sangre periférica ni de médula ósea, si bien el estudio inmunofenotípico realizado mediante citometría de flujo en el aspirado medular demostraba que el 1% de linfocitos B correspondía a una población clonal de línea B, CD5 y CD10 negativa, con restricción de cadena ligera kappa. El proteinograma demostraba una banda anómala en la zona gamma, aunque no he podido obtener la dosificación de Ig, y la inmunofijación en suero mostraba 2 bandas clonales kappa, IgM e IgG. Estos datos quizás acercarían más el diagnóstico hacia un linfoma de zona marginal, si bien este tumor, aunque también linfoma linfoplasmacítico, es muy inusual en la localización esofágica, tal como ha comentado la Dra. Galán. Posiblemente, habría sido interesante revisar la anatomía patológica de la punción pulmonar para intentar establecer un diagnóstico más concluyente. Sin embargo, las biopsias se realizaron en otro centro y no fue posible revisarlas.
Dr. Ricard Cervera. ¿Pueden comentarnos los clínicos que atendieron a la paciente cuál fue su evolución tras el diagnóstico?
Dr. Francesc Cardellach. La paciente presentó una mala evolución, y a los 5 días del diagnóstico, y previamente a iniciar el tratamiento, presentó fiebre e hipotensión que fue refractaria al tratamiento y falleció 24 h después. No se autorizó el estudio necrópsico.
Dra. M. Galán. Yo creo que esta paciente tenía metástasis en el pulmón, ya que esta extensión no es inusual en el caso del linfoma.
Diagnóstico anatomopatológico
Linfoma B de bajo grado con diferenciación plasmocelular (linfoma de zona marginal frente a linfoma linfoplasmacítico).
Agradecimientos
La Dra. M. Galán agradece a los compañeros del Comité de Patología Esofagogástrica del Hospital de Bellvitge-ICO, así como al Dr. Andia del IDI-Bellvitge, su colaboración en la preparación de la discusión del caso clínico.
