




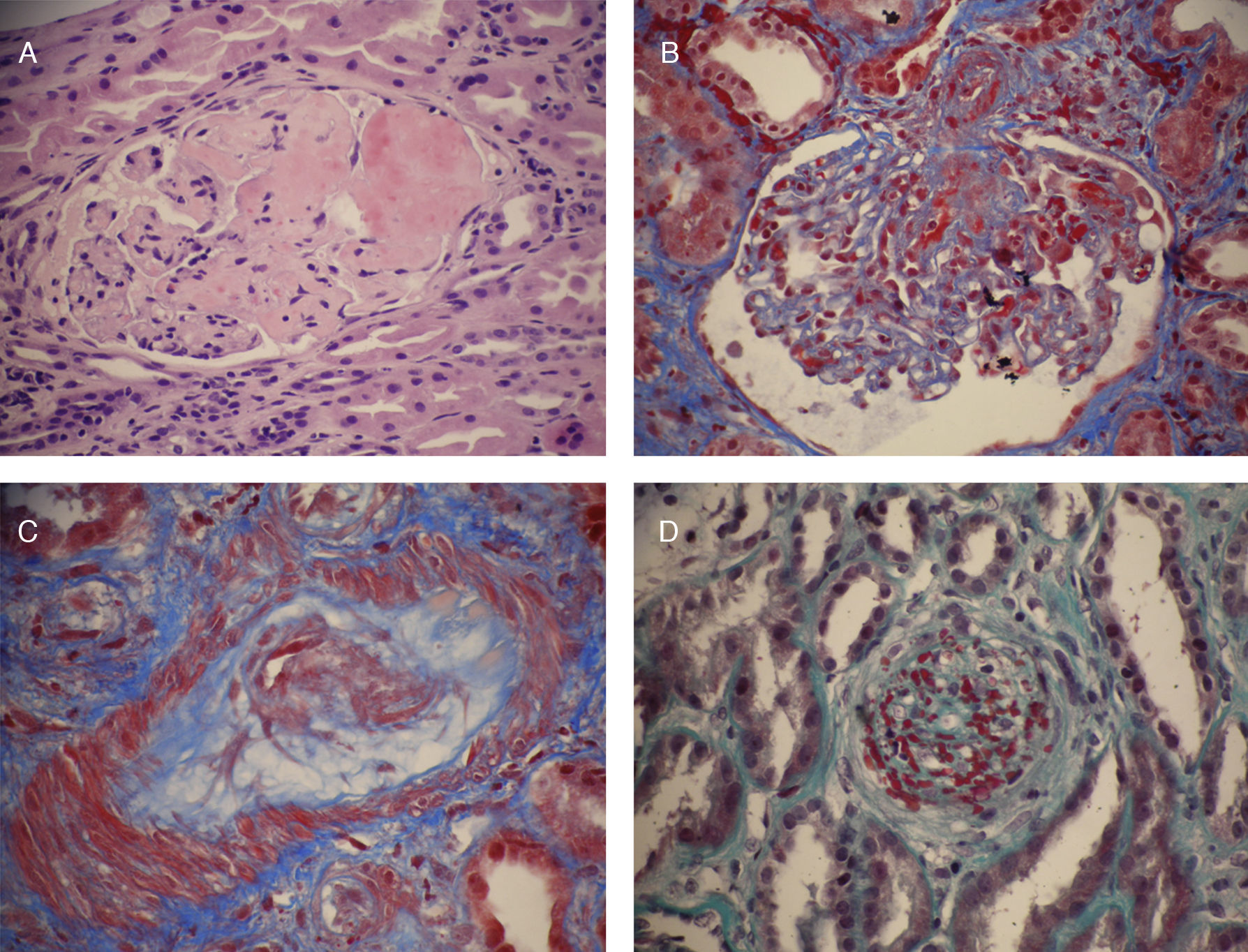
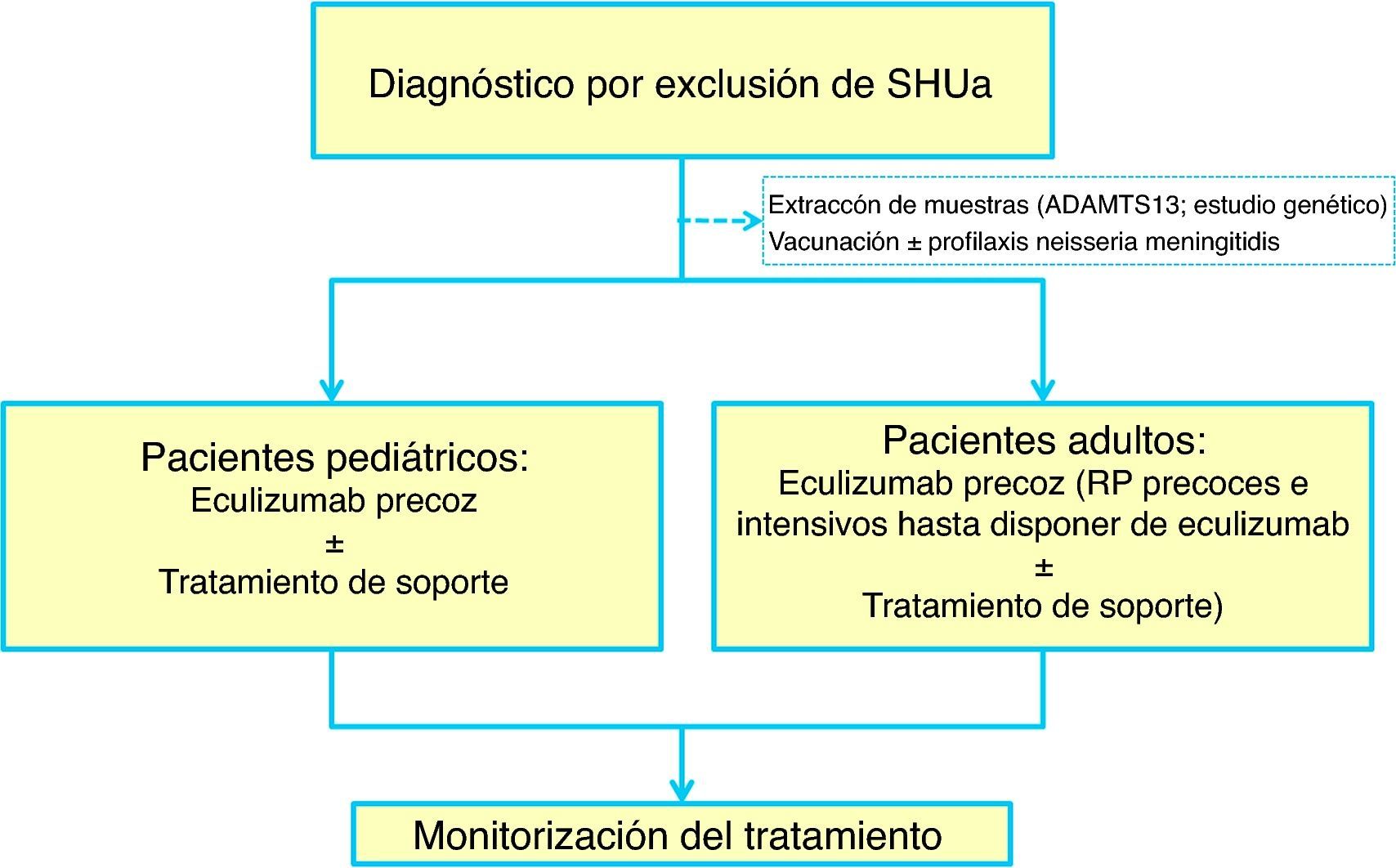
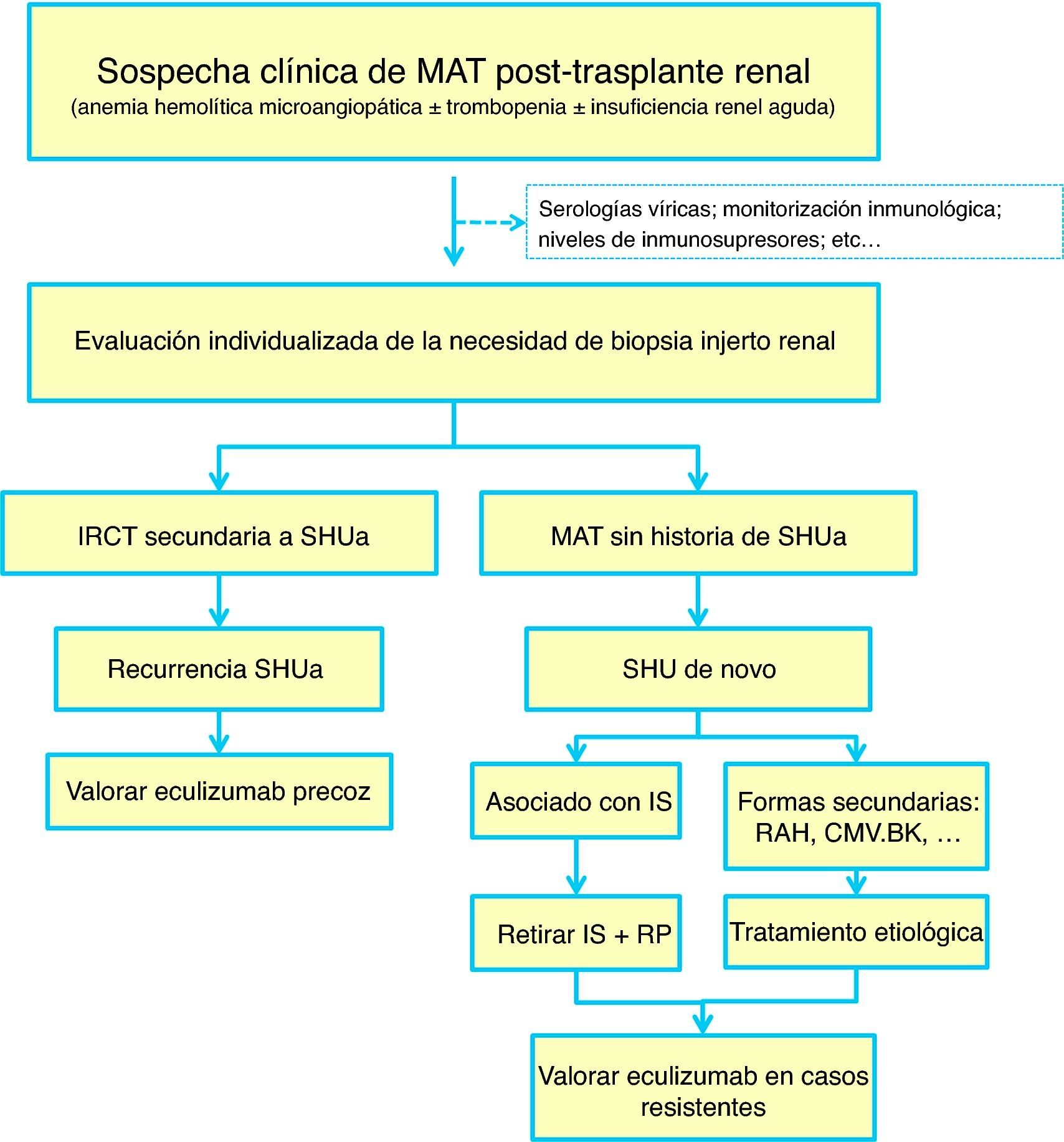
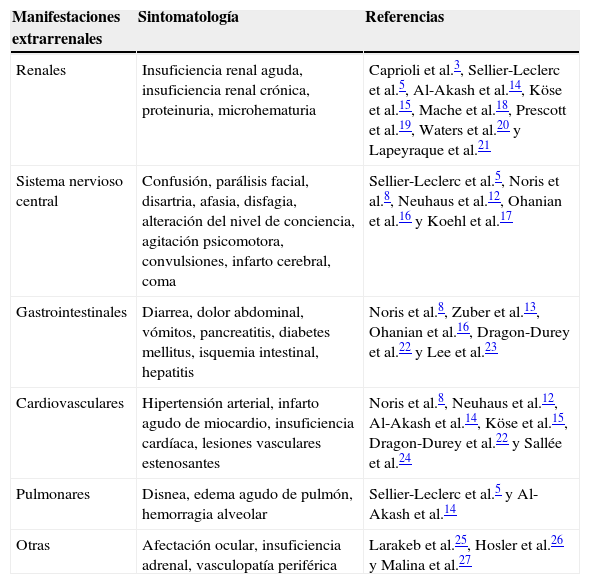



El síndrome hemolítico urémico (SHU) es una entidad clínica caracterizada por la presencia de trombocitopenia, anemia hemolítica microangiopática y daño renal, demostrando el estudio histológico la presencia de microangiopatía trombótica (MAT). Tradicionalmente el SHU ha sido clasificado en 2 formas: el SHU típico, que ocurre más frecuentemente en niños y es debido mayoritariamente a infecciones entéricas por bacterias productoras de toxina Shiga, y el SHU atípico (SHUa), que se asocia en el 50-60% de los pacientes con mutaciones en genes del sistema del complemento y que tiene un peor pronóstico, causando en la mayoría de los pacientes una insuficiencia renal crónica terminal. En el trasplante renal, el SHU se manifiesta como consecuencia de la recurrencia del SHUa o como un proceso de novo postrasplante. Durante los últimos años, numerosos estudios han puesto de manifiesto que la desregulación del sistema del complemento es la causa responsable del daño endotelial que dispara el desarrollo de la MAT en la mayoría de los pacientes con SHUa. Estos avances en el conocimiento de los mecanismos fisiopatológicos responsables del SHUa, junto con la aparición reciente de nuevas opciones terapéuticas, han permitido desarrollar mejores estrategias para conseguir un diagnóstico precoz y un tratamiento etiológico, que están cambiando la historia natural del SHUa.Esta revisión resumirá, en primer lugar, la entidad clínica del SHUa, para a continuación centrarse en la desregulación de la vía alternativa del complemento como responsable de dicha enfermedad. Finalmente, revisaremos el diagnóstico diferencial y las opciones terapéuticas del paciente con diagnóstico clínico de SHUa.
The hemolytic uremic syndrome (HUS) is a clinical entity characterized by thrombocytopenia, non-immune hemolytic anemia and renal impairment. Kidney pathology shows thrombotic microangiopathy (TMA) with endothelial cell injury leading to thrombotic occlusion of arterioles and capillaries. Traditionally, HUS was classified in 2 forms: Typical HUS, most frequently occurring in children and caused by Shiga-toxin-producing bacteria, and atypical HUS (aHUS). aHUS is associated with mutations in complement genes in 50-60% of patients and has worse prognosis, with the majority of patients developing end stage renal disease. After kidney transplantation HUS may develop as a recurrence of aHUS or as de novo disease. Over the last years, many studies have demonstrated that complement dysregulation underlies the endothelial damage that triggers the development of TMA in most of these patients. Advances in our understanding of the pathogenic mechanisms of aHUS, together with the availability of novel therapeutic options, will enable better strategies for the early diagnosis and etiological treatment, which are changing the natural history of aHUS.
This review summarizes the aHUS clinical entity and describes the role of complement dysregulation in the pathogenesis of aHUS. Finally, we review the differential diagnosis and the therapeutic options available to patients with aHUS.
Artículo

Comprando el artículo el PDF del mismo podrá ser descargado
Precio 19,34 €
Comprar ahora
