


Mujer de 27años de edad, natural de Ecuador, que acude a su médico de atención primaria con un cuadro de 48h de evolución consistente en escalofríos, tiritona y fiebre de hasta 39°C, que se acompaña de tos y expectoración espesa. Presenta dolor de características pleuríticas en el hemitórax derecho y cefalea con abundante rinorrea.
Trabaja como cuidadora de ancianos desde que hace 5meses vino a España.
Presenta una broncorrea habitual de aproximadamente 50cm3 al día, para la que no tiene seguimiento médico.
En la exploración física destaca temperatura de 37,6°C. Auscultación cardiaca rítmica, sin soplos. En la auscultación pulmonar se identifican sibilancias inspiratorias en ambos hemitórax, con crepitantes gruesos diseminados.
Hemograma: hemoglobina 12,1, hematocrito 37%, volumen corpuscular medio 89, velocidad de sedimentación globular 38, leucocitos 8.890 (41% polimorfonucleares, 36% linfocitos, 7,5% eosinófilos), plaquetas 418.000.
Bioquímica: perfiles renal, hepático, iónico y glucídico dentro de la normalidad; albúmina 3,2.
Coagulación: sin alteraciones.
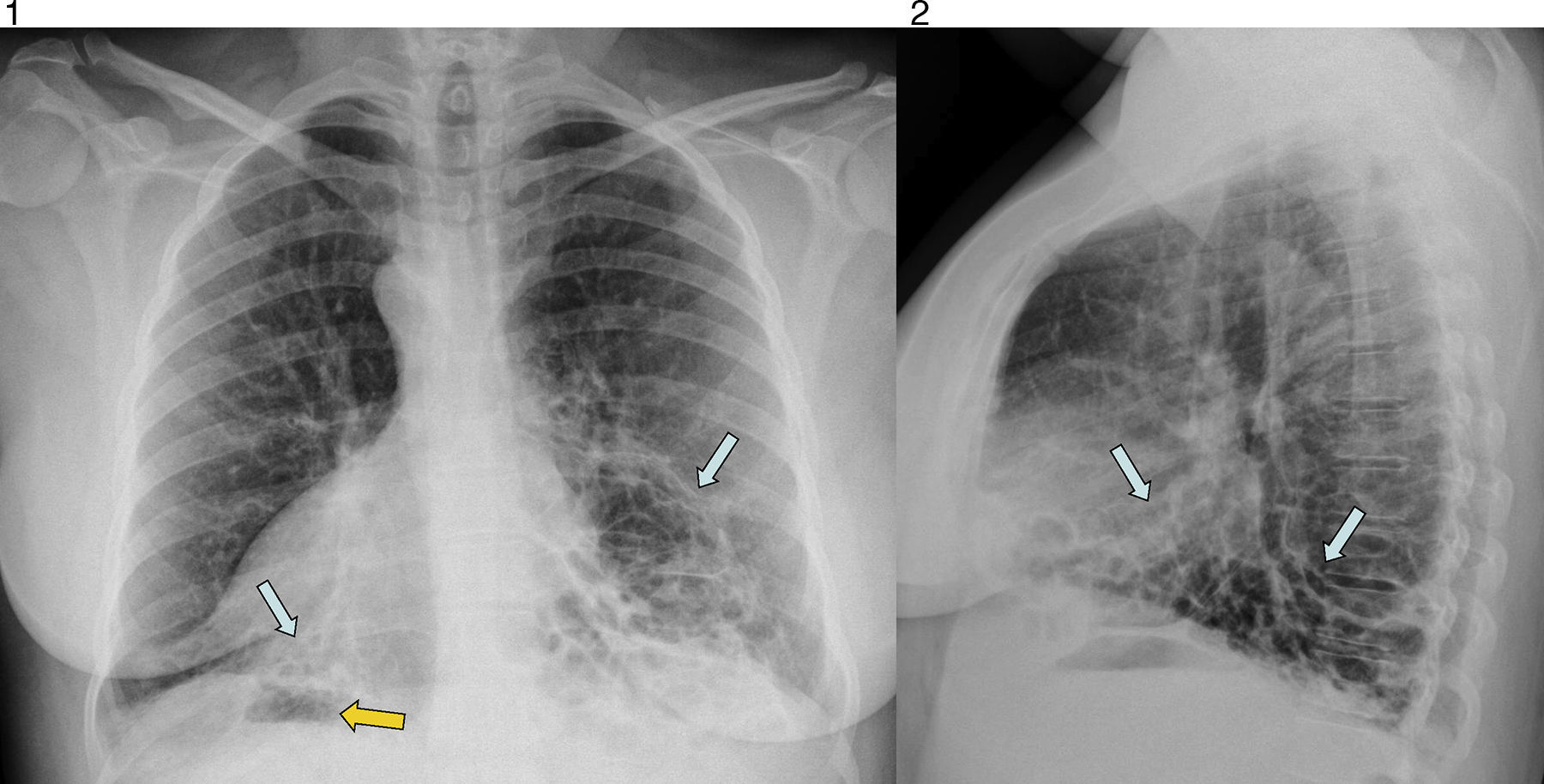
Para descartar la existencia de un foco infeccioso respiratorio, se solicitan radiografías posteroanterior y lateral del tórax. En ellas se observan bronquiectasias quísticas bibasales y dextrocardia con situs inversus: se identifica la burbuja gástrica en el lado derecho (figs. 1 y 2).
; dextrocardia con situs inversus (burbuja gástrica en el lado derecho con flecha naranja).")
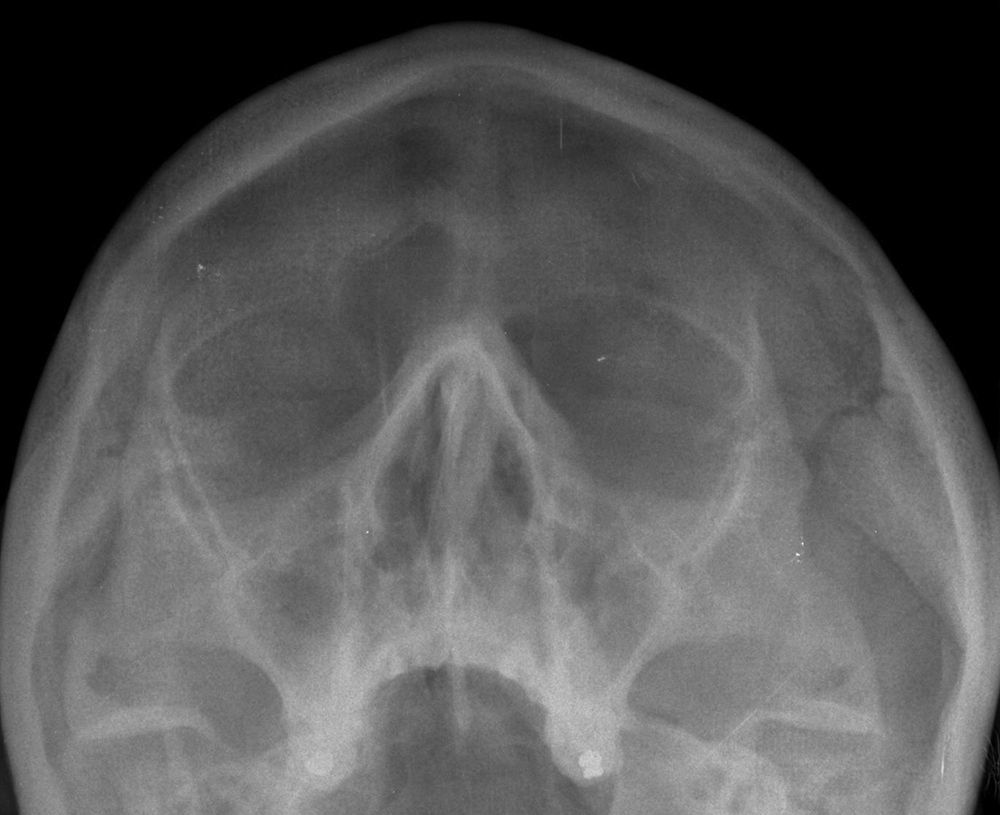
Se realiza radiografía de los senos paranasales, que muestra ocupación del seno frontal izquierdo y de ambos senos maxilares, secundaria a sinusitis (fig. 3).

Se remite a la paciente a Urgencias hospitalarias e ingresa a cargo de Neumología.
Se instaura tratamiento con oxigenoterapia, corticoides, broncodilatadores, ceftazidima y azitromicina. La clínica mejora.
El estudio se completa con tomografía computarizada (TC) torácica. En ella se evidencia dextrocardia y bronquiectasias quísticas bilaterales de gran tamaño, que afectan más al pulmón izquierdo; marcado engrosamiento de la pared y niveles hidroaéreos secundarios a infección de las mismas.
Los cultivos de esputo son positivos para Haemophilus influenzae biotipoIV.
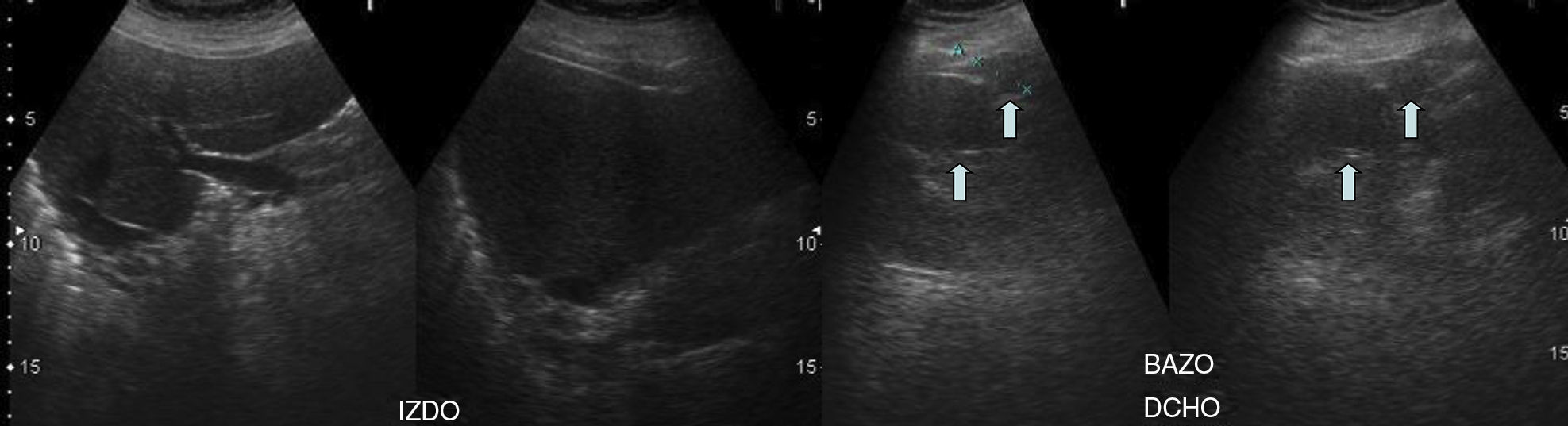
Ante la sospecha de la existencia de síndrome de Kartagener, el estudio se completa con ecografía abdominal (fig. 4), que muestra el hígado en el lado izquierdo y múltiples bazos en el lado derecho (poliesplenia).
.")
La determinación de alfa-1-antitripsina es normal.
La paciente presenta buena evolución con tratamiento médico. En el momento del alta la expectoración es más fluida; la gasometría arterial muestra pH7,42, pCO244, pO262,8, HCO328, saturación 91%.
La paciente es dada de alta con corticoides, inhaladores, expectorantes y fisioterapia respiratoria.
Comentario1-6El síndrome de Kartagener es una enfermedad genética autosómica recesiva. Fue descrita inicialmente en 1933 en 4 pacientes que presentaban la asociación de situs inversus, sinusitis de repetición y bronquiectasias.
La prevalencia estimada por estudios radiográficos y observaciones clínicas en la etapa previa al microscopio electrónico es de 1/15.000-20.000 nacidos vivos. Sin embargo, trabajos recientes que basan el diagnóstico en el estudio ultraestructural ciliar calculan una cifra mayor de la estimada previamente y la sitúan en 1/10.000 nacidos vivos.
Se considera un síndrome de discinesia ciliar primaria (DCP), que incluye un grupo de enfermedades en las que los cilios respiratorios son inmóviles (síndrome de inmovilidad ciliar), el movimiento ciliar es discinético o no hay cilios (aplasia ciliar); este último es extremadamente infrecuente.
En 1976 Afzelius describió como origen del trastorno la ausencia de brazos de dineína en los microtúbulos de los cilios bronquiales y de los flagelos de los espermatozoides.
Los cilios se encuentran desde el oído medio hasta los bronquiolos; de ahí que las manifestaciones clínicas del tracto respiratorio superior e inferior sean los puntos cardinales de la DCP.
Con frecuencia se presentan desde el nacimiento, con tendencia a la cronicidad y ausencia de respuesta a los tratamientos habituales: distrés respiratorio en neonatos sin patología que lo justifique, infecciones recurrentes de vías aéreas superiores, infecciones recurrentes de vías aéreas inferiores (más de 3 episodios con distinta localización o más de 2 episodios con idéntica localización radiológica), sinusitis recurrente, atelectasias, otitis media aguda recurrente (asociada a déficit auditivo y posible retraso del habla), otorrea, historia de asma atípica sin respuesta a tratamiento habitual, bronquiectasias no explicables por otras causas. Debido a la amplia distribución de las células ciliadas en el organismo, suelen aparecer varios síntomas en un mismo paciente.
La función pulmonar puede ser normal durante la infancia. Los datos revelan que la función pulmonar permanece relativamente estable en un porcentaje significativo de individuos, aunque suele empeorar con el tiempo.
Otras manifestaciones son el situs inversus, el hígado poliquístico, las enfermedades renales, la retinopatía, la hidrocefalia, la atresia de vías biliares, los embarazos ectópicos y la infertilidad masculina por defecto en los flagelos.
Los defectos de ultraestructura de los cilios pueden ser variados: afectar a los brazos de dineína, a las proteínas radiales, alteración en el número de microtúbulos y/o en su disposición en el axonema. Defectos mayores pueden ser la ausencia o alteraciones del axonema o de la membrana plasmática de los cilios y de los flagelos.
Si el grado de afectación es menor de 50%, las anomalías observadas pueden ser hallazgos fisiológicos o ser secundarias a infecciones víricas, inflamación de las vías aéreas o exposición a tóxicos.
Debido a la gran variedad de proteínas involucradas en el trastorno ciliar, los genes responsables también son varios y se encuentran localizados en distintos cromosomas. Actualmente se conocen al menos 10 genes relacionados con esta patología. La mayoría de los involucrados no han sido identificados, por lo que la posibilidad de un diagnóstico molecular parece todavía lejana.
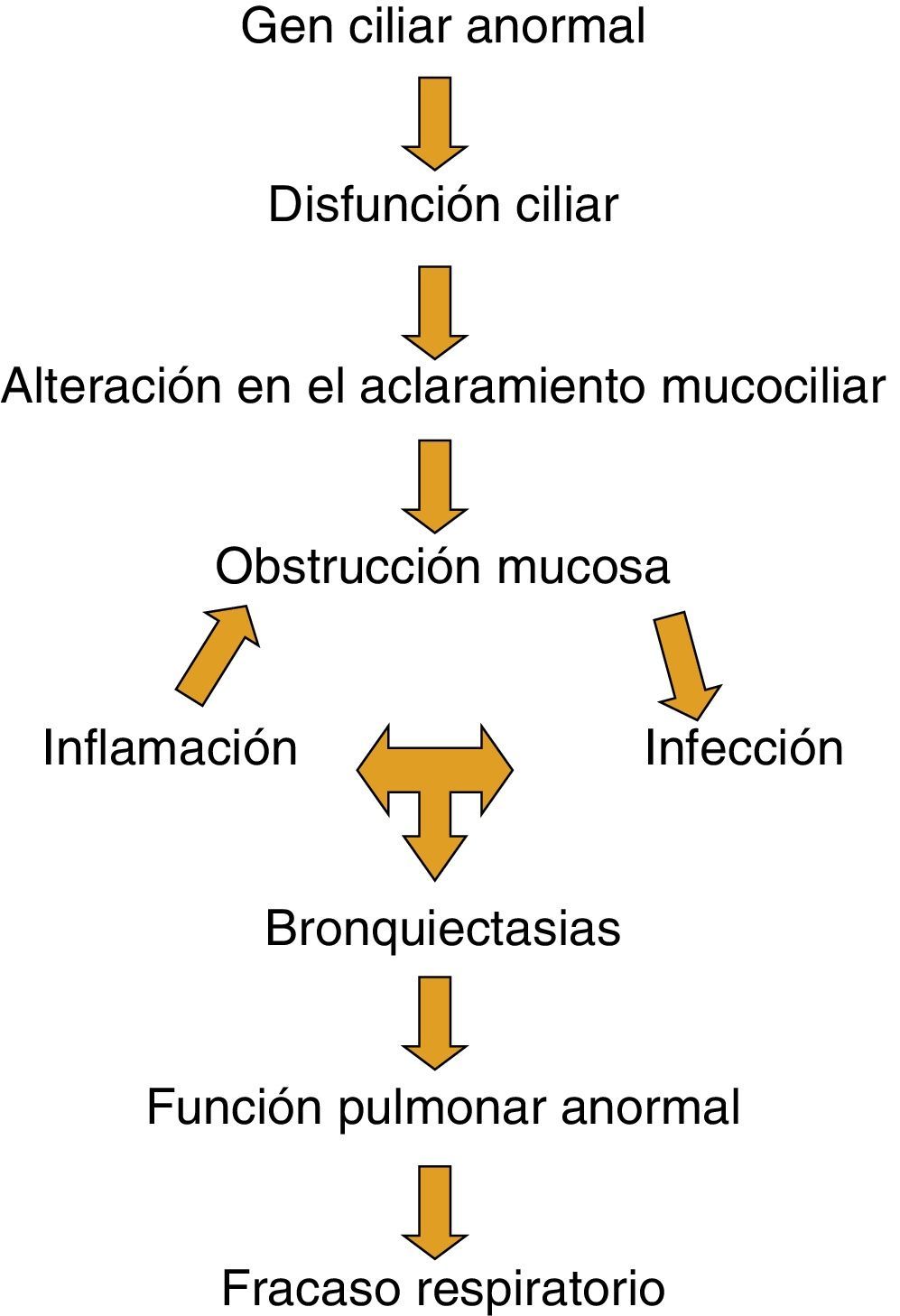
Aunque no está claro cómo el defecto básico en la DCP produce daño pulmonar y fracaso respiratorio, es reconocida la cascada de acontecimientos de la figura 5.

Si bien es un dato muy característico, el situs inversus no se asocia a todos los fenotipos de DCP. Aproximadamente el 50% de los pacientes tienen un situs inversus total debido a la ineficacia de los cilios nodales presentes de forma transitoria durante el desarrollo embrionario, lo que hace que los órganos internos se dispongan al azar.
Ante la sospecha clínica, como pruebas de cribado se utilizan diferentes técnicas:
- •
Prueba de la sacarina. Valora el aclaramiento mucociliar; su positividad indica el estudio de discinesia ciliar. Mide el tiempo de transporte mucociliar nasal. Se coloca una partícula de sacarina sódica de 1mm de diámetro en la superficie del cornete nasal inferior, 1cm por detrás de su cabeza, para evitar el epitelio escamoso; el sujeto debe permanecer sentado con la cabeza ligeramente inclinada hacia delante, con respiración fisiológica, sin estornudar ni sonarse; se le advierte de que avise cuando note algún sabor especial, sin especificar cuál es para evitar falsos positivos. Mediante el movimiento ciliar, la partícula de sacarina recorre toda la fosa nasal hasta llegar a la orofaringe, momento en que se percibe un sabor dulce característico; si tras 60min el sujeto no percibe ningún sabor, se coloca una partícula de sacarina sobre la lengua para comprobar que no tiene alteraciones del gusto. Este método fue introducido por Andersen et al. en 1974; desde entonces se ha utilizado en múltiples trabajos. La correlación entre el aclaramiento mucociliar nasal y el traqueobronquial ha quedado establecida en distintos estudios, por lo que el estudio global de la función mucociliar no necesita métodos invasivos. No es útil en niños pequeños o no colaboradores, pues deben permanecer sentados y realizar una valoración subjetiva.
- •
Prueba del óxido nítrico (ON) nasal. Consiste en detectar la disminución de ON que se produce en la DCP. La medición del ON nasal se realiza mediante quimioluminiscencia. Tras una inspiración a capacidad pulmonar total, el paciente mantiene la respiración a máxima tolerancia, se obstruye un orificio nasal y se deja permeable el otro. El analizador de ON aspira aire de forma continua de la fosa nasal obstruida. Los niveles medios de ON nasal en niños con DCP son un 90% inferiores respecto a los valores medios de niños sanos.
- •
Prueba de la microalbúmina marcada con tecnecio 99 metaestable (99mTc). Se emplea 0,01ml de macroagregados de albúmina marcados con 25 mcCi de 99mTc como trazador de radiactividad. Se depositan en la región posterior de la cabeza del cornete inferior. La prueba se efectúa a temperatura ambiente; dura 15min y después se limpia la cavidad nasal con suero fisiológico. La velocidad de transporte mucociliar nasal se calcula en función de la posición inicial y final del trazador, el tiempo consumido y la velocidad. Una velocidad de transporte mucociliar nasal mayor de 4mm/min es normal y descarta DCP. Puede llevarse a cabo incluso en recién nacidos, ya que la dosis radiactiva no supone riesgo alguno. Es una prueba objetiva, precisa y reproducible.
Las alteraciones radiológicas incluyen adelgazamiento peribronquial, atelectasias y atrapamiento aéreo, que conducen a bronquiectasias. Estos hallazgos en la radiografía de tórax típicamente aparecen en los lóbulos pulmonares medios e inferiores; es distinguible de la fibrosis quística, en la que las alteraciones pulmonares ocurren comúnmente en los lóbulos superiores.
La TC de tórax es la técnica de referencia para el diagnóstico de bronquiectasias, aunque no hay datos que apoyen la realización de TC de tórax periódicos, por la temprana edad de los pacientes y el riesgo de radiación.
Ante la sintomatología del paciente, podemos considerar otros diagnósticos que excluiremos con las pruebas específicas: la fibrosis quística (prueba del sudor), los déficits inmunitarios (con la cuantificación de inmunoglobulinas y subpoblaciones de linfocitos en sangre), la alergia respiratoria (prick-test y las IgE específicas), la aspiración pulmonar secundaria a reflujo gastroesofágico (pHmetría) o la incoordinación de la deglución (realizando videofluoroscopia deglutoria).
Estos pacientes deben ser tratados en centros especializados. La base es la fisioterapia respiratoria, la inmunización contra las enfermedades respiratorias y el control de las infecciones, para evitar el uso de antibióticos durante el mayor tiempo posible.
En algunos casos, puede ser necesario el tratamiento quirúrgico:
- •
Segmentectomía o lobectomía: limitada a pacientes seleccionados con bronquiectasias graves localizadas, en los que falla el tratamiento médico.
- •
Trasplante pulmonar: es una opción para la enfermedad pulmonar en estadios graves con afectación bilateral, empeoramiento funcional intenso, dependencia de oxigenoterapia y fallo del tratamiento médico o quirúrgico.