



CAPOS/CAOS syndrome (cerebellar ataxia, areflexia, with/without pes cavus, optic atrophy, and sensorineural hearing loss)1 is a rare autosomal dominant genetic disorder, with only 29 reported cases according to our literature search on PubMed. It is associated with heterozygous missense mutation c.2452G>A (p.Glu818Lys) of the ATP1A3 gene, and is characterised by episodes of neurological impairment associated with encephalopathy and weakness. The syndrome usually manifests at an early age following an acute episode of fever.2
We present a new case of de novo CAPOS/CAOS syndrome associated with the previously mentioned ATP1A3 mutation, and review the available literature on the topic.
Our patient is a 46-year-old woman, the first of 2 siblings (the other sibling is a 40-year-old man, who is asymptomatic); she was born to non-consanguineous parents. She had no relevant personal history. She came to our hospital due to optic atrophy with bilateral amaurosis, sensorineural hearing loss, and gait ataxia, developing progressively from childhood.
At 22 months of age, the patient had presented 3 episodes of fever within a 6-month period, with no associated infectious disease identified; she also presented partially reversible gait instability. The results of a muscle biopsy performed at the time were normal. At 5 years of age, she developed progressive hearing loss; sural nerve biopsy revealed no abnormalities. Around the age of 10 years, she presented another episode of high fever with no apparent focus of infection, associated with muscle weakness and worsening of balance, which prevented her from walking and performing fine motor tasks. She partially recovered from this episode, remaining stable until the present time. Lastly, at age 12 she presented progressive loss of visual acuity, associated with bilateral optic atrophy.
Our examination revealed bilateral anacousia and amaurosis, bilateral atrophy of the optic papilla, bilateral horizontal nystagmus with preserved saccades, mild weakness of the palpebral orbicularis muscle bilaterally, mild hypotonia predominantly in the upper limbs and paresis (4+/5) of the shoulder girdles and hips, universal areflexia, extensor plantar reflex in the right foot and slightly extensor plantar reflex in the left, mild-to-moderate upper- and lower-limb dysmetria, truncal ataxia, wide-based gait, and positive Romberg sign.
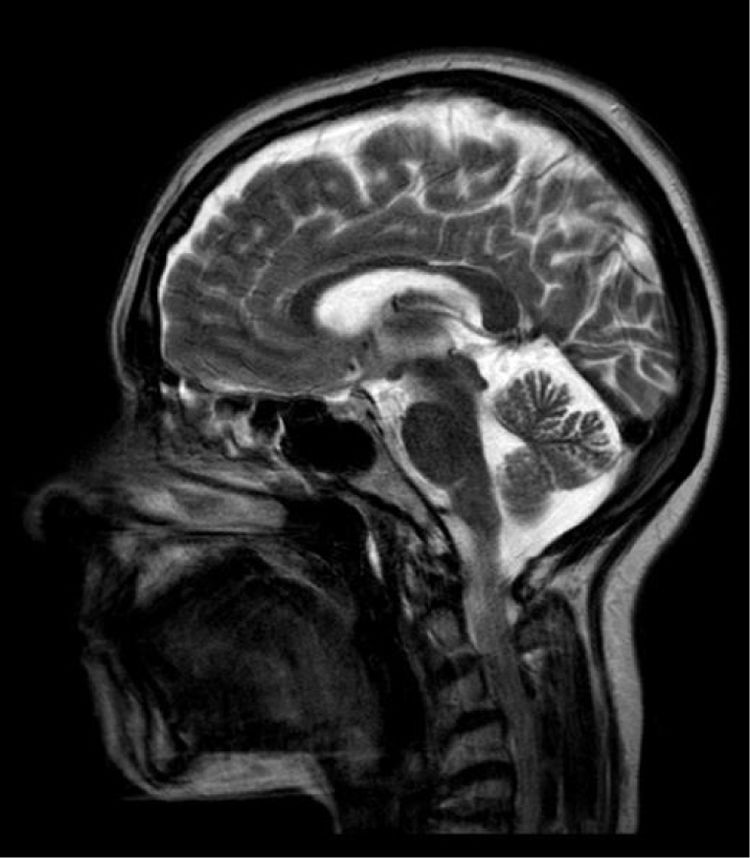
An MRI scan of the lower limbs revealed no muscle alterations, and a brain MRI scan only displayed mild cerebellar atrophy, greater than that expected given the patient's age (Fig. 1).

A neurophysiological examination including electroneurography and electromyography studies revealed no abnormal findings.
A genetic study of mitochondrial myopathies (analysis of mitochondrial DNA plus a mitochondrial and nuclear gene panel) yielded negative results. An optic atrophy panel identified a heterozygous mutation in exon 18 of the ATP1A3 gene (NM_152296): c.2452G>A; p.Glu818Lys. This mutation is described as pathogenic in the database consulted (ClinVar) and is responsible for CAPOS/CAOS syndrome, following an autosomal dominant inheritance pattern.2 The family segregation study showed that the mutation appeared de novo in our patient; this therefore constitutes a sporadic case of CAPOS/CAOS syndrome.
The patient's clinical status stabilised, and symptoms have presented no further worsening to date.
DiscussionCAPOS/CAOS syndrome (OMIM #601338) is a rare entity first described by Nicolaides et al.1 in 1996; the name of the syndrome is an acronym of the associated symptoms. In 2014, Demos and van Karnebeek3 described an association between the condition and ATP1A3 variant c.2452G>A (p.Glu818Lys) in heterozygosis.
ATP1A3 encodes ATPase Na+/K+ transporting subunit α3, which is mainly expressed in the cochlea, optic nerve, cerebellar cortex, and nerve fibres innervating muscles; this largely explains the clinical characteristics of the disease.4
In addition to CAPOS/CAOS syndrome, a rare entity compared to the classic phenotypes, other neurological diseases have been associated with mutations in the ATP1A3 gene, showing different clinical phenotypes, such as alternating hemiplegia of childhood (OMIM #614820) and rapid-onset dystonia-parkinsonism (OMIM #128235). In the last 2 years, such other phenotypes as early infantile epileptic encephalopathy, relapsing encephalopathy with cerebellar ataxia, rapid-onset ataxia, early-onset autonomic seizures, and paroxysmal asymmetric dystonic arm posturing have also been reported. However, the group of diseases associated with mutations in the ATP1A3 gene will continue to expand. Some authors have suggested that these disorders may represent a phenotypic continuum rather than distinct allele diseases.5
Symptoms typically appear during childhood. Patients present episodes of ataxia induced by fever; encephalopathy and muscle weakness appear in some cases. During this stage, patients may present encephalitis or atypical symptoms of Guillain-Barré syndrome.2 Ataxia usually improves after the acute episode, although some patients may not recover completely. All the patients described to date have presented 1-3 acute episodes.
The least consistent feature of the syndrome is pes cavus, which is absent in up to 70% of cases (as in our patient); this has led some authors to use the term CAPOS/CAOS syndrome.6 Many patients do not present signs of myopathy or neuropathy in complementary tests; this was the case with our patient.
Given the clinical heterogeneity of the disease and its overlap with other disorders, differential diagnosis should include syndromic dominant optic atrophy and other mitochondrial respiratory chain disorders.
There is no specific treatment for CAPOS/CAOS syndrome. We suggest using acetazolamide, mainly to prevent or reduce neurological symptoms during acute episodes of fever. The action mechanism of this treatment is based on the inhibition of carbonic anhydrase, decreasing extracellular pH, which seems to be associated with better Na+/K+-ATPase function.4
Please cite this article as: González Plata A, Marcos Toledano MM, Correa Martínez L, Fernández-Burriel Tercero M. Un nuevo caso de síndrome CAPOS/CAOS. Neurología. 2020;35:612–614.
