




Simultaneous presence of nuchal rigidity and skin lesions is suggestive of probable infectious aetiology. However, these signs may also be due to other causes, including drugs, tumours, and such inflammatory diseases as Sweet syndrome.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a condition of unknown aetiology that may appear isolated or in association with tumours or inflammatory or autoimmune diseases. Sweet syndrome manifests with skin lesions, systemic symptoms, and occasionally neurological symptoms.
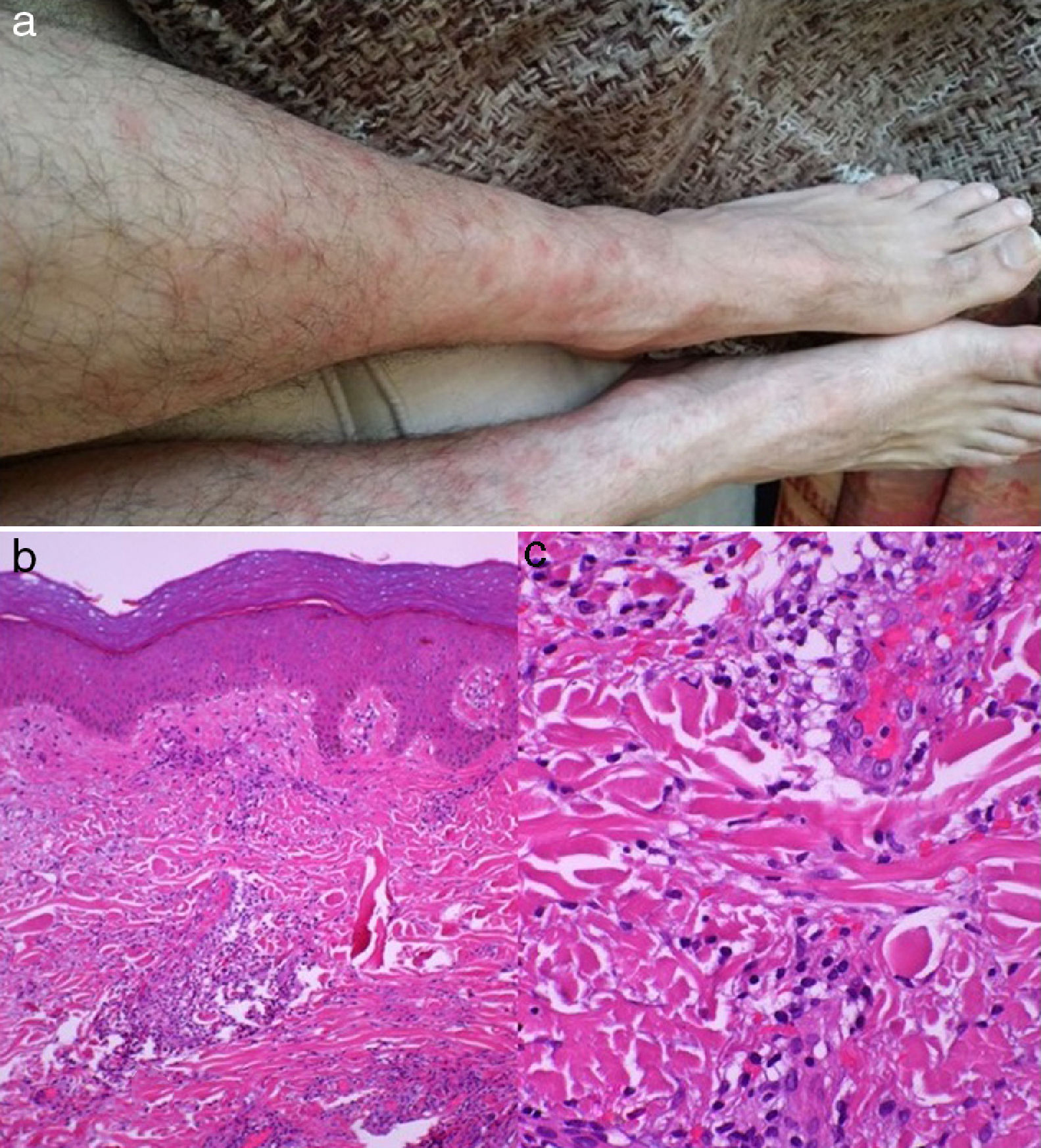
We describe the case of a 47-year-old man with no relevant medical history who came to our hospital due to a 2-week history of general discomfort, odynophagia, holocranial headache, bone and muscle pain, and fever; he was treated with amoxicillin and ibuprofen, without success. One week later, he displayed nuchal rigidity and a painful skin eruption on the chest, arms, and legs, in the form of confluent erythematous plaques and nodules.
A CSF analysis revealed pleocytosis (73cells/mm3; 100% lymphocytes), high protein levels (0.53g/L), and normal glucose levels. Bacterial cultures and polymerase chain reaction for herpes virus, enterovirus, Streptococcus, Listeria monocytogenes, and Neisseria meningitidis yielded negative results. A blood analysis revealed an erythrocyte sedimentation rate of 94mm/h, a C-reactive protein level of 4.82mg/dL (normal range, ≤0.5mg/dL), leukocytosis (12700 leukocytes/mm3, 78% neutrophils), anticardiolipin IgM 21.91U/mL (normal range, ≤12.5U/mL), anti-β2 glycoprotein 1 (B2GP1) IgM 104.51U/mL (normal range, ≤20U/mL), and normal levels of tumour markers. A skin biopsy showed neutrophil infiltration in the dermis, and no involvement of blood vessel walls or microthrombosis (Fig. 1). A chest and abdomen CT scan, an MRI study, and a gastroscopy revealed no abnormal findings. No association was found with HLA-Cw1 or HLA-B54.
. Skin biopsy: haematoxylin-eosin stain showing intradermal neutrophil infiltration with no vascular involvement or microthrombosis (b and c).")
The patient received ceftriaxone plus aciclovir; these drugs were subsequently discontinued due to the absence of bacterial or viral infection. Oral methylprednisolone dosed at 75mg/day improved skin eruptions and neurological signs. Six months later, tests for anti-B2GP1 IgM and IgG yielded negative results.
Once antimicrobial drugs were discontinued and other aetiologies ruled out; our patient was diagnosed with Sweet syndrome with neurological involvement in view of the lack of vascular involvement in the skin biopsy and the excellent response to corticosteroids.
Sweet syndrome is a rare inflammatory disease presenting with a wide range of manifestations. It is characterised by sudden-onset oedema, painful erythematous plaques or nodules on the skin, fever, and leukocytosis associated with neutrophilia.1 In addition to the skin, the condition may also involve the respiratory system, bones and muscles, and the central nervous system (CNS).
From a histopathological viewpoint, Sweet syndrome is characterised by neutrophil infiltration into the dermis, with no vascular invasion or signs of infection. Occasionally, cells are predominantly mononuclear, mainly immature granulocytes misinterpreted as histiocytes (histiocytoid Sweet syndrome).2
Sweet syndrome has been associated with numerous diseases, and can be classified as: (1) classic Sweet syndrome, (2) Sweet syndrome associated with neoplasms, or (3) drug-induced Sweet syndrome.
Classic Sweet syndrome accounts for the majority of cases; diagnosis is made after ruling out an association with malignant tumours or adverse drug reactions. The condition may be associated with infections, inflammatory bowel disease, pregnancy, or autoimmune disease.3
Cohen et al.4 reviewed the published cases of 79 patients with Sweet syndrome associated with malignancies; 87% were associated with blood cancer (lymphoma, chronic leukaemia, myeloma, and myelodysplastic syndromes), and the remaining cases with solid tumours. Another study by the same researchers included 118 cases of Sweet syndrome, 21% of which were associated with solid tumours or blood cancer.5
Sweet syndrome has been associated with the administration of multiple drugs, the most frequent being the granulocyte colony stimulating factor (Table 1).3
Drugs associated with Sweet syndrome.
| Antibiotics | Clindamycin, cyclins (doxycycline, minocycline, and tetracycline), nitrofurantoin, quinolones (norfloxacin and ofloxacin), piperacillin/tazobactam, streptogramins (dalfopristin/quinupristin), trimethoprim-sulfamethoxazole |
| Antivirals | Abacavir, aciclovir, interferon alpha |
| Biotherapeutic products | Proteasome inhibitors (bortezomib), tyrosine kinase inhibitors (imatinib, nilotinib), granulocyte colony stimulating factors, all-trans retinoic acid, macrophage colony-stimulating factor |
| NSAIDs | Celecoxib, rofecoxib, diclofenac |
| Psychotropic drugs | Amoxapine, clozapine, diazepam, lormetazepam |
| Vaccines | Tuberculosis (Bacillus Calmette-Guérin vaccine), Streptococcus pneumoniae (Pneumovax®), influenza, smallpox |
| Miscellaneous | Azathioprine, radiocontrast agents, propylthiouracil, carbamazepine, chloroquine, furosemide, hydralazine, isotretinoin, lenalidomide, oral contraceptives |
The pathogenic mechanisms of Sweet syndrome are unknown. It has been hypothesised that the condition may be caused by a genetic predisposition in HLA carriers (Cw1 and B54), cytokine dysregulation, and hypersensitivity reactions.
CNS involvement, in the form of meningitis or encephalitis, and other neurological complications has been extensively reported in the literature. Hisanaga et al.6 described 42 cases of Sweet syndrome with neurological involvement, proposing a set of diagnostic criteria for neuro-Sweet disease.
A patient with probable neuro-Sweet disease should meet the following criteria: (1) neurological symptoms rapidly responding to corticosteroids; (2) painful plaques or nodules on the face, neck, trunk, or upper limbs, associated with neutrophil infiltration into the dermis; (3) absence of uveitis or cutaneous vasculitis (unlike in Behçet syndrome); and (4) absence of other causes of CNS involvement. Our patient met these diagnostic criteria.
Neuro-Sweet disease is usually associated with good prognosis. Patients respond favourably to systemic corticosteroids, although some may experience recurrences.7 Differential diagnosis must include any cause of lymphocytic meningoencephalitis, especially Behçet syndrome.8
Antiphospholipid antibodies have been associated with multiple autoimmune diseases; they are present in up to 5% of the population, although only a small percentage of these individuals develop vascular complications.9 The presence of anti-B2GP1 antibodies in a patient with Sweet syndrome is a previously unreported finding.
The fact that the anti-B2GP1 antibodies disappeared and were not converted to IgG antibodies, the resolution of cutaneous and neurological manifestations after corticosteroid treatment, and the lack of vascular or neoplastic phenomena after 18months of follow-up support the hypothesis that this case of classic Sweet syndrome with neurological manifestations was autoimmune in origin.
Please cite this article as: Cruz-Velásquez GJ, Pac Sha J, Simal Gil E, Gazulla J. Meningitis aséptica y anticuerpos anti-β2-glicoproteína-1 en el síndrome de Sweet. Neurología. 2018;33:620–621.
This study was presented in poster format at the 66th Annual Meeting of the Spanish Society of Neurology, held in Valencia in November 2014.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas