





Steinert's disease or myotonic dystrophy type 1 (MD1), (OMIM 160900), is the most prevalent myopathy in adults. It is a multisystemic disorder with dysfunction of virtually all organs and tissues and a great phenotypical variability, which implies that it has to be addressed by different specialities with experience in the disease. The knowledge of the disease and its management has changed dramatically in recent years. This guide tries to establish recommendations for the diagnosis, prognosis, follow-up and treatment of the complications of MD1.
Material and methodsConsensus guide developed through a multidisciplinary approach with a systematic literature review. Neurologists, pulmonologists, cardiologists, endocrinologists, neuropaediatricians and geneticists have participated in the guide.
RecommendationsThe genetic diagnosis should quantify the number of CTG repetitions. MD1 patients need cardiac and respiratory lifetime follow-up. Before any surgery under general anaesthesia, a respiratory evaluation must be done. Dysphagia must be screened periodically. Genetic counselling must be offered to patients and relatives.
ConclusionMD1 is a multisystemic disease that requires specialised multidisciplinary follow-up.
La enfermedad de Steinert o distrofia miotónica tipo 1 (DM1), (OMIM 160900) es la miopatía más prevalente en el adulto. Es una enfermedad multisistémica con alteración de prácticamente todos los órganos y tejidos y una variabilidad fenotípica muy amplia, lo que implica que deba ser atendida por diferentes especialistas que dominen las alteraciones más importantes. En los últimos años se ha avanzado de manera exponencial en el conocimiento de la enfermedad y en su manejo. El objetivo de la guía es establecer recomendaciones para el diagnóstico, el pronóstico, el seguimiento y el tratamiento de las diferentes alteraciones de la DM1.
Material y métodosEsta guía de consenso se ha realizado de manera multidisciplinar. Se ha contado con neurólogos, neumólogos, cardiólogos, endocrinólogos, neuropediatras y genetistas que han realizado una revisión sistemática de la literatura.
RecomendacionesSe recomienda realizar un diagnóstico genético con cuantificación precisa de tripletes CTG. Los pacientes con DM1 deben seguir control cardiológico y neumológico de por vida. Antes de cualquier cirugía con anestesia general debe realizarse una evaluación respiratoria. Debe monitorizarse la presencia de síntomas de disfagia periódicamente. Debe ofrecerse consejo genético a los pacientes con DM1 y a sus familiares.
ConclusiónLa DM1 es una enfermedad multisistémica que requiere un seguimiento en unidades especializadas multidisciplinares.
Myotonic dystrophy type 1, Steinert disease, Steinert–Curschmann disease, Batten–Gibb disease, atrophic myotonia, DM1, or MD1 (OMIM 160900, ORPHA 273, ICD-9-CM 359.21, ICD-10 G71.1, ICD-11 8C71.0) is the most prevalent myopathy in adults.1,2 It is an autosomal dominant disease caused by a CTG trinucleotide repeat expansion in the noncoding region of the DMPK gene (myotonic dystrophy protein kinase), located on the long arm of chromosome 19 (19q13.3). The disease is traditionally diagnosed by neurologists due to its characteristic neuromuscular alterations, although it also causes systemic alterations. It is considered to be one of the diseases with the greatest phenotypic variability. As a result of the variability of its manifestations, both in terms of quality and quantity, every patient requires personalised treatment and clinicians must be well aware of the possibility of alterations affecting each organ and system in order to ensure that patients receive the most appropriate treatment and follow-up.
MethodsThese guidelines are intended for reference by professionals involved in the diagnosis and follow-up of patients with MD1. They include specific recommendations for the diagnosis, follow-up, and treatment of alterations associated with the disease.
The working group that developed these guidelines includes specialists from all disciplines involved in the care of patients with MD1: neurology, pulmonology, cardiology, endocrinology, paediatric neurology, and genetics. The guidelines take into account the perspectives and preferences of patients and their families and clearly define the intended users.
The guidelines are based on a literature search of the Cochrane Library, Cochrane Plus, EMBASE, PubMed-MEDLINE, and ECA LOST databases and follow the PICO (patient, intervention, comparison, outcome) method recommended by the working group for clinical practice guidelines of the Spanish national healthcare system.3 The search terms used were “myotonic dystrophy,” “myotonic dystrophy type 1,” and “Steinert's disease”; no date range was specified. We also used bibliographical references from the articles identified. We selected the most recent and most rigorous articles published in English, French, and Spanish.
The working group was divided into subgroups to review the articles, with one group addressing each of the following topics: diagnosis; motor and cognitive alterations; cardiac alterations; respiratory alterations; other alterations; and MD1 in paediatric patients. Each subgroup drafted an initial document, and an ad hoc meeting was held at the spring meeting of the Spanish Society of Neurology's Neuromuscular Disorders Study Group, in Santiago de Compostela on 29 April 2017, where these documents were debated and agreed. The definitive version of the guidelines was agreed in subsequent discussions.
The authors represent the national expert committee on MD1, an interdisciplinary panel of physicians with extensive clinical experience with the disease. All authors approved the final version of this manuscript and take full responsibility for its content.
The AGREE II tool4 was used to evaluate and guarantee the quality, clarity, rigour, applicability, and editorial independence of these guidelines.
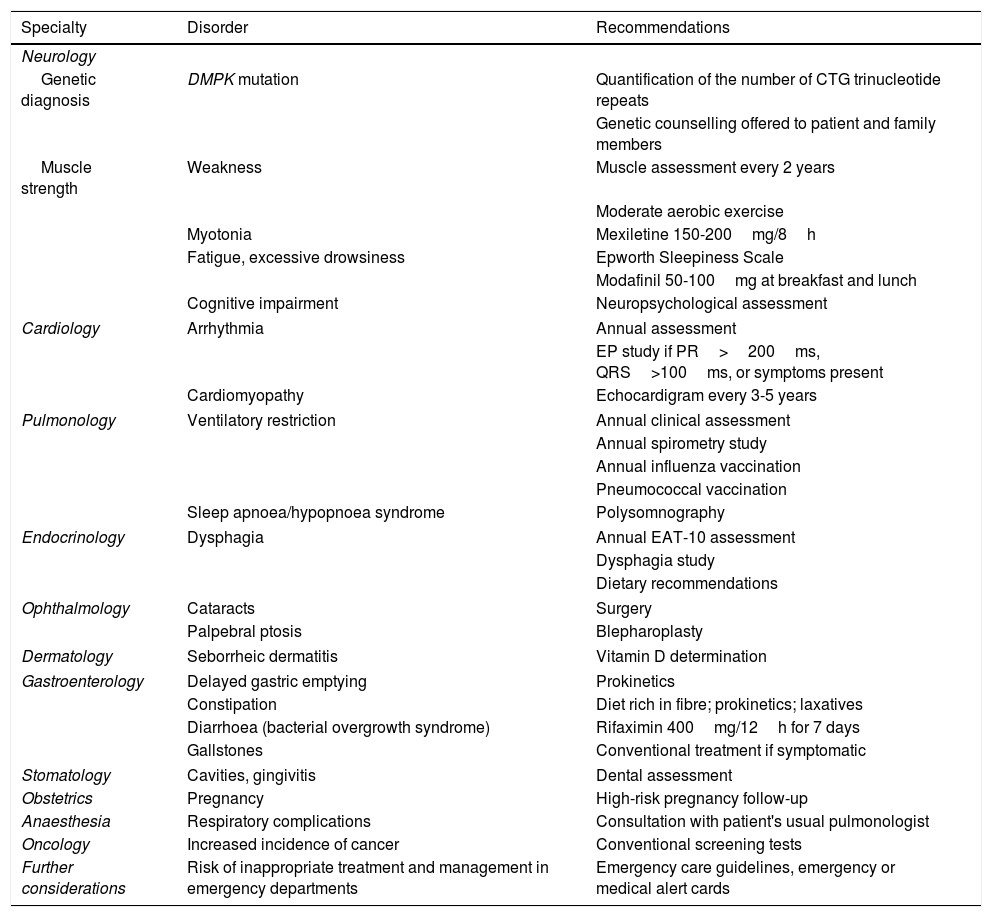
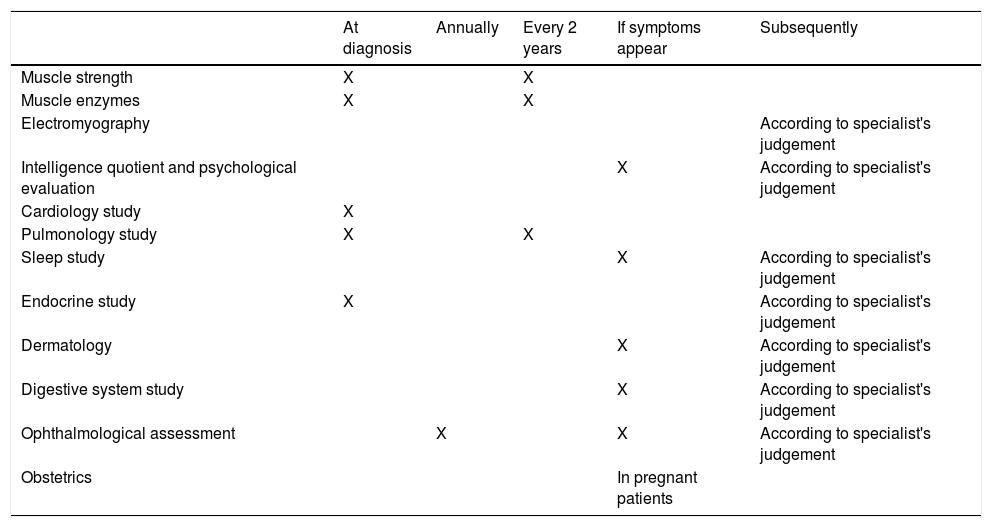
Table 1 summarises the recommendations made and Table 2 shows the recommended intervals between evaluations.
Summary of the consensus recommendations.
| Specialty | Disorder | Recommendations |
|---|---|---|
| Neurology | ||
| Genetic diagnosis | DMPK mutation | Quantification of the number of CTG trinucleotide repeats |
| Genetic counselling offered to patient and family members | ||
| Muscle strength | Weakness | Muscle assessment every 2 years |
| Moderate aerobic exercise | ||
| Myotonia | Mexiletine 150-200mg/8h | |
| Fatigue, excessive drowsiness | Epworth Sleepiness Scale | |
| Modafinil 50-100mg at breakfast and lunch | ||
| Cognitive impairment | Neuropsychological assessment | |
| Cardiology | Arrhythmia | Annual assessment |
| EP study if PR>200ms, QRS>100ms, or symptoms present | ||
| Cardiomyopathy | Echocardigram every 3-5 years | |
| Pulmonology | Ventilatory restriction | Annual clinical assessment |
| Annual spirometry study | ||
| Annual influenza vaccination | ||
| Pneumococcal vaccination | ||
| Sleep apnoea/hypopnoea syndrome | Polysomnography | |
| Endocrinology | Dysphagia | Annual EAT-10 assessment |
| Dysphagia study | ||
| Dietary recommendations | ||
| Ophthalmology | Cataracts | Surgery |
| Palpebral ptosis | Blepharoplasty | |
| Dermatology | Seborrheic dermatitis | Vitamin D determination |
| Gastroenterology | Delayed gastric emptying | Prokinetics |
| Constipation | Diet rich in fibre; prokinetics; laxatives | |
| Diarrhoea (bacterial overgrowth syndrome) | Rifaximin 400mg/12h for 7 days | |
| Gallstones | Conventional treatment if symptomatic | |
| Stomatology | Cavities, gingivitis | Dental assessment |
| Obstetrics | Pregnancy | High-risk pregnancy follow-up |
| Anaesthesia | Respiratory complications | Consultation with patient's usual pulmonologist |
| Oncology | Increased incidence of cancer | Conventional screening tests |
| Further considerations | Risk of inappropriate treatment and management in emergency departments | Emergency care guidelines, emergency or medical alert cards |
Recommended periodic assessments.
| At diagnosis | Annually | Every 2 years | If symptoms appear | Subsequently | |
|---|---|---|---|---|---|
| Muscle strength | X | X | |||
| Muscle enzymes | X | X | |||
| Electromyography | According to specialist's judgement | ||||
| Intelligence quotient and psychological evaluation | X | According to specialist's judgement | |||
| Cardiology study | X | ||||
| Pulmonology study | X | X | |||
| Sleep study | X | According to specialist's judgement | |||
| Endocrine study | X | According to specialist's judgement | |||
| Dermatology | X | According to specialist's judgement | |||
| Digestive system study | X | According to specialist's judgement | |||
| Ophthalmological assessment | X | X | According to specialist's judgement | ||
| Obstetrics | In pregnant patients |
Creatine kinase levels may be slightly elevated in patients with the typical forms of the disease but are usually normal in asymptomatic individuals.
Neurophysiological studyElectromyography studies show typical myotonic discharges, consisting of trains of positive waves or fibrillation potentials of waning amplitude and frequency; the audio profile resembles a dive bomber aeroplane. The electromyography pattern shows characteristic myopathic traits with polyphasic, low-amplitude potentials and an early interference pattern. While these alterations may be observed in any muscle, distal involvement is more evident.
Electroneurography shows a decrease in motor evoked potential amplitude.
Short-exercise testing reveals an early fall in evoked motor potential amplitude immediately after effort, similar to that observed in channelopathies secondary to alterations to the chloride channel gene (Thomsen-type and Becker-type myotonia). This is as we would expect, given that myotonia in MD1 is thought to be caused by an alteration in the transcription of the gene encoding the chloride channel (CLCN-1), which is mutated in congenital myotonia. This finding helps to differentiate MD1 from MD2 (proximal myotonic myopathy or PROMM); in the latter condition, motor evoked potential amplitude is not modified with effort.
Muscle biopsyMuscle biopsy has never been considered an essential procedure in the diagnosis of MD1, and the disease presents no pathognomonic histopathological characteristics. However, the association of a large number of centrally located nuclei, aggregations of pyknotic nuclei, sarcoplasmic masses, ring fibres or moth-eaten fibres, and selective atrophy of type 1 fibres is very suggestive of MD1. These findings are similar to those observed in MD2, with the difference that type 2 fibre atrophy is predominant in the latter condition.
In any case, the availability and specificity of genetic diagnosis make muscle biopsy unnecessary in cases of suspected MD1.
Muscle magnetic resonance imagingAlthough muscle magnetic resonance imaging (MRI) has become a fundamental diagnostic procedure in the study of neuromuscular diseases, little experience with the technique has been published in the study of MD1 due to the variable expressivity of its clinical features and the accessibility of genetic testing. In the upper limbs, some studies report involvement of the flexor digitorum profundus, flexor digitorum superficialis, flexor pollicis longus, extensor pollicis brevis and longus, and abductor pollicis brevis muscles, the lateral head of the triceps brachii, and the infraspinatus. In the lower limbs, the tibialis anterior is initially affected, with subsequent involvement of the semimembranosus and vastus intermedius muscles and the medial head of the gastrocnemius. Early involvement of the paravertebral muscles may also be detected. Good correlation is reported between clinical expression and alterations on muscle MRI.
Genetic diagnosisDiagnostic confirmation of MD1 requires detection of the genetic alteration responsible for the disease. The mutation involved is a CTG trinucleotide repeat expansion in a noncoding region at the 3′ end of the DMPK gene, located on the long arm of chromosome 19 (19q13.3).
Healthy individuals have between 5 and 34 CTG trinucleotide repeats. Individuals with 35 to 49 repeats (premutation) present no symptoms of the disease but can transmit a greater number of repeats to their offspring, who may reach the mutation range. Individuals presenting over 50 repeats are considered to have the mutant allele and do present symptoms; disease severity is correlated with the number of trinucleotide repeats. Generally, greater numbers of CTG repeats are associated with earlier onset and more numerous and more severe symptoms. This genotype-phenotype correlation is stronger below 400 repeats. When more than 400 repeats are present, the instability of mitotic transmission causes errors in transmission of the fragment between parent and daughter cells, resulting in genetic mosaicism, with different tissues presenting different numbers of expansions. As a result, the number of trinucleotide repeats detected in lymphocytes may not be proportional to the number of repeats in other tissues, such as in muscles. In fact, the number of CTG repeats in skeletal muscle is usually 2 to 13 times greater than that measured in leukocytes.
Patients with 50-100 CTG repeats typically present milder forms of the disease, developing cataracts at 50-60 years of age and/or mild myotonia. Patients with the classic form usually have 100-1000 CTG repeats, with severity increasing in line with the number of repeats. Finally, most patients with congenital myotonic dystrophy have expansions of over 1000 repeats. Nonetheless, this correlation is not perfect, and there are patients whose symptoms are more or less severe than we may expect given the number of repeats detected. Furthermore, the size of the trinucleotide repeat expansion may change with age; therefore, the most accurate genotype-phenotype correlation is observed when genetic testing is performed close to the time of clinical assessment.
Detection of CTG repeat expansionsThe CTG repeat expansion is studied with an ethylenediamine tetraacetic acid test of a 10-mL blood sample. Determining the size of the expansion in other tissues (e.g., muscle) does not inform diagnosis.
Polymerase chain reaction (PCR) testing is used to detect expansions of up to 180 trinucleotide repeats. Triple-repeat primed PCR is an inexpensive technique that detects whether the number of CTG repeats is within the normal or the pathological range, but cannot precisely quantify repeats. Southern blot techniques are needed to determine the number of CTG repeats above 180.
This molecular study detects practically 100% of pathogenic variants; therefore, the sensitivity and specificity of the genetic study approach 100%. Detecting whether the number of repeats is greater or smaller than 50 is insufficient. Quantifying CTG repeats is essential, given the prognostic value of this information.
Genetic counselling. AnticipationMD1 follows an autosomal dominant inheritance pattern. Therefore, the likelihood of the mutation being transmitted to offspring is 50%. Penetrance is very high, approaching 100% in individuals aged 50 years if all clinical manifestations are taken into account.
The severity of the phenotype in the child depends on the size of the CTG fragment inherited. As transmission of the fragment is unstable, with a tendency for the number of trinucleotide repeats to be greater in the offspring, children inheriting the mutation tend to present more severe forms than their parents; this phenomenon is known as genetic anticipation. Expansions of between 50 and 80 CTG repeats may be transmitted for several generations without any great changes. However, alleles of this size are less stable when they are transmitted by the father. Beyond this range, transmission by the mother is usually associated with greater differences between generations, and the increase in the number of CTG repeats can be great enough to reach the range associated with congenital MD1. The risk of the child having congenital MD increases in line with the size of the repeat expansion in the mother, especially if over 300 CTG repeats are present; however, there is always a risk of the mother passing the mutation to her offspring, even with smaller expansions. It is even possible for a woman who is asymptomatic or presents very mild symptoms to have a child with the congenital variant.
It is very rare for the transmitted CTG fragment to be shorter in the child. This phenomenon is more frequent when the gene is inherited from a father with larger fragments (greater than 500 CTG repeats).
Whenever a patient is diagnosed with MD1, family members should be made aware of the risk of developing the disease, even if they are currently asymptomatic, and genetic studies should be proposed to those aged over 18 years. Presymptomatic diagnosis is considered inappropriate in minors as no treatment is available for the disease and diagnosis denies the individual autonomy over what they do or do not wish to know, risks stigmatisation and discrimination, and may lead to anxiety in the child or overprotection on the part of parents. The need for appropriate genetic counselling is especially pressing in women of childbearing age who are at risk, given the possibility that their children may present congenital forms of the disease. In any case, early detection of all carriers of the mutation is very important on account of the risk of severe complications, particularly cardiac complications. Before these tests are performed, it is advisable to inform patients about the characteristics and transmission of the disease.
Family planning. Prenatal and pre-implantation genetic diagnosisIt is possible to diagnose the disease in embryos and even to select healthy embryos in vitro for subsequent implantation. These options should be analysed and offered prior to pregnancy.
Prenatal diagnosis involves genetic testing of a chorionic villus sample extracted between 9 and 12 weeks of gestation. It can also be diagnosed by amniocentesis in week 16. These procedures are associated with a risk of miscarriage of 3%-4%.
Pre-implantation genetic diagnosis may significantly reduce the risk of miscarriage, making it a relevant technique in the context of a disease associated with reduced fertility and an elevated risk of miscarriage. Furthermore, it represents a way of bypassing the ethical issues associated with therapeutic abortion. Nonetheless, the procedure requires ovarian stimulation and oocyte retrieval, which are very demanding for patients. The procedure should be started before the ovarian reserve decreases either due to natural causes (around the age of 38 years) or because of the disease itself. The success rate of in vitro fertilisation is relatively low and it is often necessary to repeat the procedure several times. Furthermore, as a chorionic villus sample is often needed to confirm absence of the mutation in the embryo, the risk of miscarriage is the same as that associated with prenatal diagnosis.
Diagnostic protocolIn cases of suspected MD1, the diagnostic procedure of choice is a genetic study. The other techniques mentioned may be of interest from a research viewpoint, but are not required to establish a diagnosis. Furthermore, normal results in those tests, even the electromyography study, do not rule out presence of the disease.
Even in families with no known history of the disease, we should suspect MD1 and consider performing genetic tests in the child and parents if the neonate presents hypotonia or if second- or third-trimester ultrasound studies show evidence of polyhydramnios or a lack of foetal movement.
Because of the complexity of the disease, patients with MD1 should be followed up at neuromuscular disorders units with experience managing the condition and with access to numerous specialties. Every specialist involved in the care of the patient should have good understanding of MD1.
Neurological involvementMuscular symptomsWeaknessWeakness in MD1 shows clear distal predominance, although the muscles of the neck and face and those involved in mastication, swallowing, and speech are also affected. Weakness progresses slowly, at an estimated rate of 1%-3% per year.5 Patients present elevated risk of falls and fractures.
Several scales are used to quantify weakness:
- i)
The Muscular Impairment Rating Scale6 is a validated instrument in which muscle weakness is assessed manually in 11 muscle groups, on a scale of 1 (no muscular impairment) to 5 (severe proximal weakness). The muscle groups evaluated are:
- a.
Neck flexors
- b.
Shoulder abductors
- c.
Elbow flexors
- d.
Wrist extensors
- e.
Digital flexors
- f.
Hip flexors
- g.
Knee extensors
- h.
Knee flexors
- i.
Ankle dorsiflexors
- j.
Ankle plantar flexors
- a.
- ii)
Medical Research Council scale for muscle strength
- iii)
Hand grip dynamometry
- iv)
Functional tests7:
- •
Six-minute walk test (distance)
- •
Ten-metre walk test (timed)
- •
Four-step stair up (timed)
- •
Four-step stair down (timed)
- •
Rising from the floor (timed)
- •
- v)
Questions about:
- •
Limitations in walking
- •
Falls
- •
Limitations with transfers
- •
Use and convenience of wheelchairs
- •
Aerobic exercise and strength training of moderate intensity do not cause any damage, although their effectiveness is yet to be demonstrated.8 Moderate exercise is recommended for all patients.5 Excessive exercise may increase the rate of disease progression, and is not advised. It has been demonstrated that patients with MD1 fall and stumble up to 10 times more frequently than the general population,9 with the associated risk of bone fractures; the use of ankle-foot orthoses may prevent these accidents.10
MyotoniaMyotonia is a disorder of muscle relaxation after voluntary contraction, which patients usually describe as stiffness; myotonia improves with heat and repeated exercise, and increases with cold and resting. This symptom is very frequent, but causes few symptoms and does not always require treatment. It may favour the appearance of muscle rigidity, dysarthria, dysphagia, pain, and gastrointestinal symptoms.
TreatmentMexiletine dosed at 100-200mg 3 times daily has been demonstrated to be effective in treating myotonia associated with MD1 (class I evidence).11 Mexiletine and other antiarrhythmic drugs should be administered with caution due to their ability to block sodium channels in the heart (which presents functional alterations in MD1), acting on the excitability of these channels and consequently affecting cardiac arrhythmias and atrioventricular conduction.12–15
Other treatments include phenytoin, carbamazepine, clomipramine, imipramine, amitriptyline, nifedipine, flecainide, acetazolamide, and taurine. As with mexiletine, caution should be exercised when prescribing other drugs.16
MyalgiaPain is a very frequent symptom of MD1, reported in up to 90% of patients; clinicians should always enquire about pain and treat it with appropriate analgesics.17
TreatmentPregabalin may be tried at low doses (50-75mg/day).
FatigueFatigue is a frequent symptom of MD1 and lacks a clear relationship with muscle weakness.18 No validated Spanish-language scales are available, but the Fatigue Severity Scale may be used.19
TreatmentModafinil (200mg/day) is recommended in the first week, and the dose may be doubled if the desired effect is not achieved.20 This is an off-label indication.
Central nervous system disordersCentral nervous system involvement can be one of the greatest problems affecting the daily lives of patients with MD1. These manifestations are highly variable and include cognitive deficits, apathy, fatigue, and sleep disorders; patients with the congenital form may present intellectual disability, attention-deficit/hyperactivity disorder, and executive dysfunction.21,22
Intellectual disabilityIntellectual disability is very evident in congenital forms of MD1, but may also present with varying severity in other forms, although some patients present a normal intelligence quotient.23 Patients with MD1 tend to have a lower level of schooling and lower socioeconomic level than controls, particularly among men.24
Progressive memory loss has also been demonstrated, even in patients with relatively small trinucleotide repeat expansions.25,26
There is currently no general consensus on which tests should be used to study cognitive deficits in MD1. A screening battery has been proposed,24 including the following tests: (1) an abbreviated form of the Wechsler Adult Intelligence Scale (WAIS)27 to quantify intelligence quotient, or the corresponding test for children: the Wechsler Intelligence Scale for Children or Weschler Preschool and Primary Scale of Intelligence; (2) the Rey Auditory Verbal Learning Test to evaluate verbal memory; (3) the Rey Complex Figure Test to explore visual memory; and (4) the Digit Span and Block Design subtests of the WAIS-IV to assess attention and information processing speed. These tests are affected by several potential confounding factors, such as weakness, myotonia, fatigue, and depression. Therefore, they should only be used in select cases or for academic studies.
Patients with the congenital form often present language disorders, intellectual disability, neurocognitive dysfunction, attention deficits, hyperactivity, or autism spectrum disorders.28,29
It has recently been suggested that cognitive behavioural therapy may improve fatigue and participation in social activities in patients with MD1.30
Sleep disordersSleep disorders can be the reason for patients’ first consultation. One-third of patients present excessive daytime sleepiness, particularly after eating21,31; patients have difficulty remaining awake, and periodic limb movements have been reported during sleep.32 Sleep apnoea/hypopnoea syndrome (SAHS) is also very frequent. However, excessive daytime sleepiness does not show a strong association with SAHS and does not always respond to treatment.
The Epworth Sleepiness Scale is a good instrument for diagnosing these disorders.33
Depression and personality disordersWhile psychiatric problems are usually not severe, major depression and personality alterations can result in specific psychological characteristics in patients with MD1.34 They often show traits of schizoid disorder, anxiety, hysteria, compulsion, depressive neurosis, narcissistic or self-destructive behaviour, lack of initiative, and apathy.24,35,36
Depression is usually evaluated with the Hamilton Depression Rating Scale.37
Cerebrovascular diseaseThe incidence of stroke among patients with MD1 is not well defined, but has been associated with cardioembolism secondary to arrhythmia.38–40
NeuroimagingCentral nervous system disorders seem to show some association with histopathological abnormalities41 and neuroimaging findings. For example, reduced grey matter volume and diffuse white matter disruption have been found to be correlated with the size of the CTG repeat expansion.21,42–44
FDG-PET imaging may show fronto-temporal hypometabolism bilaterally, although a correlation with neuropsychological deficits has not been demonstrated.42
Cardiac involvementCardiac involvement is a frequent, significant manifestation of MD1. Between 75% and 80% of patients are estimated to be affected to some extent, with a variable clinical spectrum: symptoms range from mild electrocardiographic (ECG) alterations to severe arrhythmias that may cause sudden death. In fact, cardiac causes account for up to one-third of mortality in these patients.
A Danish registry of 1186 patients with MD1 found arrhythmias, cardiomyopathy, and cardiac insufficiency to be particularly frequent.45 The pathophysiology of these disorders may be explained by fibrofatty replacement of the specialised tissue responsible for cardiac and muscular conduction in both ventricles.46–48
No clear relationship has been demonstrated between cardiac events and the number of CTG repeats or neuromuscular involvement, and the evidence on the subject is contradictory.47 For this reason, and because the risk of cardiac alterations is high from a young age,49 it is essential to ensure early detection of these complications through lifelong follow-up, even in patients with no or little muscular involvement.
Conduction disorders, arrhythmia, and sudden deathThe most frequent cardiac manifestations of MD1 are ECG alterations and arrhythmias. The most affected part of the cardiac conduction system is the His-Purkinje system, although other areas are also affected (e.g., the sinoatrial or atrioventricular nodes).5,6 According to a recent meta-analysis, the most frequent disorders are first-degree atrioventricular block (28.2%), prolonged QT interval (22%) or QRS complex duration (19.9%), ventricular extrasystole (14.6%), left bundle branch block (5.7%), atrial fibrillation/flutter (5%), right bundle branch block (4.4%), and nonsustained ventricular tachycardia (4.1%).47
Arrhythmias can cause palpitations, syncope, and sudden death; clinically asymptomatic patients may also present arrythmias. The risk of sudden death in patients with MD1 is estimated at 0.56% per year.47 Studies into the mechanisms underlying this phenomenon report contradictory results. Traditionally, ventricular fibrillation and asystole due to bradyarrhythmia have been cited as the main causes of death. It is known that asymptomatic patients with MD1 and electrophysiological (EP) study results showing a His-ventricular (HV) interval of ≥ 70ms present an elevated risk of developing third-degree atrioventricular block, and benefit from prophylactic pacemaker implantation.50 However, there seem to be other causes of sudden death that are not preventable with a pacemaker.50 Tachyarrhythmias, and particularly ventricular tachyarrhythmias, have been reported as a cause of sudden death.51
Groh et al.52 identify history of atrial tachyarrhythmia and presence of severe ECG alterations (second or third degree atrioventricular block, PR interval ≥ 240ms, and QRS duration ≥ 120ms) as predictors of sudden death. More recently, a retrospective study of a cohort of patients with MD1 showing ECG alterations at baseline (PR > 200ms and/or QRS duration > 100ms) found that an invasive strategy comprising an EP study and prophylactic pacemaker implantation for patients with HV interval ≥ 70ms was associated with a higher survival rate during the follow-up period than a non-invasive strategy.53
Myocardial involvementBetween 7% and 20% of patients with MD1 are reported to present hypertrophy, dilated cardiomyopathy, and systolic dysfunction of the left ventricle.47,53 Older age, male sex, conduction alterations on ECG, and arrhythmias are predictors of systolic dysfunction.54 This entity, which is usually asymptomatic, increases the risk of sudden death. Increased prevalence of left ventricular noncompaction and myocardial fibrosis, detectable by delayed myocardial enhancement on MRI studies, are also described.55 Patients with MD1 are also reported to present reduced myocardial mass and dysfunction of the right ventricle, as well as increased prevalence of mitral valve prolapse. Finally, we should also be aware of alterations of myocardial relaxation, which are equivalent to the myotonia affecting skeletal muscle and may present as symptoms of cardiac insufficiency.48
Recommendations56,57 (Table 3):
- 1.
Given the progressive nature of the disease, patients with MD1 should undergo lifelong cardiological follow-up, even in the absence of symptoms or evidence of cardiac involvement.
- 2.
Patients with MD1 should be informed about the alarm symptoms (syncope, palpitations) for which they should seek emergency consultation.
- 3.
A targeted clinical history interview (with special emphasis on syncope and/or palpitations), ECG, and Holter monitoring study should be performed annually.
- 4.
A transthoracic echocardiogram should be performed every 3-5 years.
- 5.
Management of structural cardiac alterations should follow a similar approach to that applied in the general population.
- 6.
Asymptomatic patients with significant ECG alterations (PR interval > 200ms; QRS duration > 100ms) or history of cardiogenic syncope may benefit from an invasive EP study to measure conduction times and the risk of ventricular arrhythmia induction. If EP study findings are normal, clinicians should consider repeating the study during follow-up if new symptoms appear or if existing ECG alterations significantly progress (an increase of > 20ms in PR interval and/or QRS duration).
- 7.
Patients with ECG findings showing HV interval > 70ms or second degree atrioventricular block benefit from prophylactic implantation of a definitive pacemaker, due to the high risk of progression to complete atrioventricular block. Pacemakers do not completely eliminate the risk of sudden death, which may be caused by other mechanisms (e.g., ventricular arrhythmia, asystole).
- 8.
Implantable cardioverter defibrillators should be considered in patients with MD1 and history of ventricular arrhythmia and/or left ventricular dysfunction. EP studies should be performed in patients with sustained monomorphic ventricular tachycardia; if bundle branch reentrant tachycardia is detected, it should be treated with catheter ablation.
- 9.
Management of supraventricular arrhythmia is similar in patients with MD1 and in other patient groups. However, special precautions should be taken with the use of antiarrhythmic drugs that may prolong the HV interval (especially class I drugs).
- 10.
Sedation of patients with MD1 for electric cardioversion should be performed with caution by trained personnel in a suitable environment, due to the risk of respiratory failure.
Cardiology recommendations.
| 1. Even in the absence of symptoms or study findings suggesting cardiac involvement, patients with MD1 should undergo lifelong cardiological follow-up to screen for possible complications, given the progressive nature of the disease. |
| 2. Patients should be informed about alarm symptoms (syncope, palpitations) for which they should urgently seek medical attention. |
| 3. A targeted clinical history interview (with special emphasis on syncope and palpitations), ECG, and Holter monitoring study should be performed annually. |
| 4. A transthoracic echocardiogram should be performed every 3-5 years. |
| 5. Structural cardiac alterations should be managed as in the general population. |
| 6. Asymptomatic patients with significant ECG alterations (PR interval > 200ms; QRS duration > 100ms) or history of cardiogenic syncope may benefit from an invasive EP study to measure conduction times and the risk of ventricular arrhythmia induction. If EP study findings are normal, clinicians should consider repeating the study during follow-up if new symptoms appear or if the existing ECG alterations progress (an increase of > 20ms in PR interval and/or QRS duration). |
| 7. Patients with ECG findings showing HV interval > 70ms or second-degree atrioventricular block benefit from prophylactic implantation of a definitive pacemaker, due to the high risk of progression to complete atrioventricular block. Pacemakers do not completely eliminate the risk of sudden death, as it may be caused by other mechanisms (e.g., ventricular arrhythmia, asystole). |
| 8. Implantable cardioverter defibrillators should be considered in patients with MD1 and history of ventricular arrhythmia and/or left ventricular dysfunction. EP studies should be performed in patients with sustained monomorphic ventricular tachycardia; if bundle branch reentrant tachycardia is detected, it should be treated with catheter ablation. |
| 9. Management of supraventricular arrhythmia is similar in patients with MD1 and in other patient groups. However, special precautions should be taken with the use of antiarrhythmic drugs that may prolong the HV interval (especially class I drugs). |
| 10. Sedation of patients with MD1 for electric cardioversion should be performed with caution by trained personnel in a suitable environment, due to the risk of respiratory failure. |
Respiratory involvement is common in patients with MD1 and constitutes one of the main causes of premature death in this patient group, accounting for 51%-75% of mortality.58,59 Furthermore, it is one of the factors with the greatest influence on quality of life.1 There seems to be a degree of correlation between the size of the CTG repeat expansion and the severity of respiratory involvement.60,61 Some studies have found that male patients present more risk factors for respiratory insufficiency.62 Obesity, which is common in patients with MD1, has been shown to be an independent risk factor for respiratory failure.63
In the majority of cases, onset is insidious and progressive, which frequently results in diagnostic delay as symptoms go undetected; it occurs late (50-60 years of age) and in patients with an established diagnosis of MD1 presenting muscular and multisystemic symptoms.64 However, cases have been reported of acute-onset respiratory insufficiency triggered by a range of processes, including anaesthesia and respiratory infections.65
The pathophysiological mechanisms causing respiratory alterations in these patients are not well understood, although many studies support a dual mechanism comprising peripheral (dystrophy and myotonia of the muscles involved in respiration [diaphragm and abdominal and intercostal muscles]) and central nervous system involvement (central nervous system anomalies affecting central respiratory control).66,67
Respiratory involvement in MD1 may be associated with ventilatory restriction due to muscle weakness, or presence of SAHS or nocturnal hypoventilation. The most frequent clinical signs suggestive of respiratory involvement are recurrent respiratory infections, progressive dyspnoea, excessive daytime sleepiness, and morning headache.66,68–71
Behavioural and cognitive alterations in patients with MD1 may constitute an obstacle for proper identification of respiratory symptoms and for adherence to nocturnal ventilation therapy.72
Ventilatory restriction due to weakness of respiratory muscles may result in hypoventilation, initially only at night.
Weakness of the intercostal and abdominal muscles increases the risk of respiratory infections due to dysphagia and reduced cough capacity. The risk of pneumonia is further increased by the elevated risk of bronchoaspiration secondary to oropharyngeal and oesophageal weakness.73,74
Given the clinical and pathophysiological heterogeneity of respiratory involvement in MD1, screening for this complication requires protocolised studies.
Recommendations (Table 4):
- 1.
Respiratory dysfunction should be assessed periodically in patients with MD1.
- 2.
Respiratory history-taking should address symptoms of dysphagia, cough effectiveness, dyspnoea, excessive daytime sleepiness, and morning headache and fatigue. The Epworth Sleepiness Scale is recommended.33
- 3.
The initial study includes spirometry, plethysmography, maximum inspiratory and expiratory pressure, sniff nasal/inspiratory force, peak expiratory cough flow, baseline gasometry, and nocturnal cardiorespiratory polygraphy (PGR).
- 4.
Nocturnal polysomnography is recommended in patients with excessive daytime sleepiness, normal or pathological PGR results, and lack of response to nocturnal ventilation therapy.
- 5.
Continuous positive airway pressure is recommended in patients with moderate or severe SAHS who do not present nocturnal hypoventilation (cumulative time spent with SpO2 below 90% [CT90]>30%).
- 6.
Non-invasive mechanical ventilation (bilevel positive airway pressure) is recommended in patients with predominant nocturnal hypoventilation (CT90>30%).
- 7.
To improve treatment adherence, patients should receive initial training in the use of nocturnal ventilation devices, on either an inpatient or outpatient basis.
- 8.
Early antibiotic treatment is recommended in the event of respiratory infection.
- 9.
Respiratory physiotherapy techniques and mechanical insufflator–exsufflator cough assist devices (peak expiratory cough flow < 270) are recommended for secretion management.
- 10.
Influenza vaccine should be administered annually and pneumococcal vaccine should be administered at least once in the patient's lifetime.
- 11.
A respiratory evaluation should be performed prior to any surgical procedure under general anaesthesia.
Pulmonology recommendations.
| 1. Respiratory dysfunction should be assessed periodically in patients with MD1. |
| 2. Respiratory history-taking should address symptoms of dysphagia, cough effectiveness, dyspnoea, excessive daytime sleepiness, and morning headache and fatigue. The Epworth Sleepiness Scale is recommended.33 |
| 3. The initial study includes spirometry, plethysmography, maximum inspiratory and expiratory pressure, sniff nasal/inspiratory force, peak expiratory cough flow, baseline gasometry, and nocturnal cardiorespiratory polygraphy (PGR). |
| 4. Nocturnal polysomnography is recommended in patients with excessive daytime sleepiness, normal or pathological PGR results, and lack of response to nocturnal ventilation therapy. |
| 5. Continuous positive airway pressure is recommended in patients with moderate or severe SAHS who do not present nocturnal hypoventilation (cumulative time spent with SpO2 below [CT90]>30%). |
| 6. Non-invasive mechanical ventilation (bilevel positive airway pressure) is recommended in patients with predominant nocturnal hypoventilation (CT90>30%). |
| 7. To improve treatment adherence, patients should receive initial training in the use of nocturnal ventilation devices, on either an inpatient or outpatient basis. |
| 8. Early antibiotic treatment is recommended in the event of respiratory infection. |
| 9. Respiratory physiotherapy techniques and mechanical insufflator–exsufflator cough assist devices (peak expiratory cough flow < 270) are recommended for secretion management. |
| 10. Influenza vaccine should be administered annually and pneumococcal vaccine should be administered at least once in the patient's lifetime. |
| 11. A respiratory evaluation should be performed prior to any surgical procedure under general anaesthesia. |
MD1 can affect multiple tissues and organs, with varying severity.
DermatologyMultiple pilomatricomas (calcifying epithelioma of Malherbe)Calcifying epithelioma of Malherbe is a benign tumour derived from matrix cells of the hair follicles; it manifests in the form of subcutaneous nodules of 0.5-5cm diameter, which are not painful. The condition affects the scalp, face, neck, and upper limbs. It can be treated with surgical resection if patients report local discomfort or aesthetic problems and histopathological diagnosis is confirmed. These growths may be mistaken for simple sebaceous cysts.75,76
AlopeciaAlopecia has multiple causes, including accelerated ageing of the skin and hair follicles, as well as hormonal changes. No specific treatment recommendations have been made.77
Seborrheic dermatitisThis condition is associated with reduced serum vitamin D levels. It is diagnosed by dermatological examination and serum vitamin D determination. Calcifediol may improve symptoms.6
Dysplastic nevus without associated melanomaThis condition is associated with reduced serum vitamin D levels. Diagnosis requires dermatological examination and serum vitamin D determination. The condition may improve with calcifediol.77
Endocrinology and metabolismPatients with MD1 may present various endocrine disorders, with the most frequent being hypogonadism, thyroid disorders, and disorders of hydrocarbon and phosphocalcic metabolism. Less frequently, patients may present corticotropic axis dysfunction, dyslipidaemia, and electrolyte alterations. Given the increase in the incidence of endocrine manifestations as the disease progresses, periodic screening is important.78
Hypergonadotropic hypogonadismHypergonadotropic hypogonadism is the most common endocrine manifestation of MD1. Testicular atrophy is reported in up to 80% of male patients, with tubular damage being more frequent than interstitial involvement. This condition is characterised by low testosterone levels and/or infertility due to altered spermatogenesis and elevated gonadotropin levels (follicle-stimulating hormone [FSH] and luteinising hormone [LH]). Testosterone deficit causes progressive regression of secondary sexual characteristics (body hair, libido, frequency of shaving) and loss of muscle mass. This loss of lean body mass may in turn exacerbate the underlying muscular symptoms of the disease, eventually causing a reduction in bone mass.
Women with the condition have a higher risk of infertility, miscarriage, and in rare cases, premature ovarian failure, which is also a cause of secondary osteoporosis.79
Given the high prevalence of hypogonadism, determination of FSH, LH, and testosterone levels should be requested at the first consultation with adult male patients, and repeated annually or sooner if they present symptoms of hypogonadism.
In women with secondary amenorrhoea, oestradiol and LH/FSH levels should be determined.
Male patients with hypogonadism should be referred to the endocrinology department for further study and evaluation of whether to start testosterone replacement therapy.
Hormone replacement therapy should also be considered in women, depending on the age of ovarian failure onset.
Disorders of hydrocarbon metabolismInsulin resistance generally constitutes the pathophysiological basis of hydrocarbon metabolism alterations in MD1, with muscles losing sensitivity to the hormone. This may present as:
- -
Hyperinsulinism with normal glucose tolerance: elevated insulin levels with normal plasma glucose concentration.
- -
Prediabetes, with altered baseline glycaemia (fasting plasma glucose of 100-125mg/dL), glycosylated haemoglobin (HbA1c) of 5.7%-6.4% (standardised method of the Diabetes Control and Complications Trial-National Glycohemoglobin Standardisation Programme [DCCT-NGSP]), or intolerance to carbohydrates (2-hour post-75g oral load glycaemia of 140-199mg/dL).
- -
Diabetes mellitus, diagnosed in patients with fasting plasma glucose ≥ 126mg/dL, post-75g oral load glycaemia ≥ 200mg/dL, or HbA1c≥ 6.5%.
Diagnosis of diabetes or prediabetes requires the presence of at least 2 criteria, except in patients with random plasma glucose > 200mg/dL and clinical symptoms (polyuria, polydipsia, polyphagia, weight loss).
Fasting plasma glucose, HbA1c, and insulin levels should be determined once annually, or sooner in patients presenting the cardinal symptoms of hyperglycaemia: polyuria, polydipsia, polyphagia, and weight loss.
The treatment of choice for patients with diabetes is insulin-sensitising drugs; metformin is the drug of first choice.
Lipid profile alterationsThe most frequent lipid profile alterations are elevated triglyceride levels (67% of patients) and decreased high-density lipoprotein levels (35%). Dyslipidaemia and hydrocarbon metabolism alterations increase cardiovascular risk due to the metabolic syndrome; therefore, arterial blood pressure and lipid profile (cholesterol, cholesterol fractions, and triglycerides) should be tested once or twice annually.
It is essential to emphasise healthy diet and regular physical exercise, adapted for each patient, given the great significance of sedentary lifestyles in the aetiopathogenesis of metabolic syndromes. In patients requiring pharmacological treatment for dyslipidaemia, statins and fibrates may be used, although close supervision is needed as these drugs may exacerbate myopathy in these patients. If these drugs are prescribed, creatine kinase, glutamic oxaloacetatic transaminase, and glutamic pyruvic transaminase levels should be monitored periodically, particularly in the first year.80
Phosphocalcic metabolismSome series report vitamin D deficiency (25-hydroxy vitamin D < 30ng/mL) in up to 90% of patients with MD1. Severe deficiency (25-hydroxy vitamin D < 10ng/mL) is detected in 40% of patients. Hypovitaminosis D has the same causes in these patients as in the general population: low intake of foods rich in vitamin D, limited exposure to sunlight, and changes in body composition (increased body fat). Vitamin D deficiency can cause osteomalacia (insufficient bone mineralisation), muscle weakness, and secondary hyperparathyroidism, which in turn worsens the existing muscle weakness in patients with myotonic dystrophy.
With regard to phosphocalcic metabolism, increased prevalence is reported of primary hyperparathyroidism (up to 17.5%), which is generally associated with parathyroid adenoma.78
Primary hyperparathyroidism is managed in the same way in patients with MD1 as in the general population.
Calcium, phosphate, vitamin D, and parathyroid hormone levels should be determined annually. If hypercalcaemia, hypophosphataemia, or elevated parathyroid hormone concentration are detected, a second test should be requested, and the patient should be referred to the endocrinology department if the alteration is confirmed.
Thyroid metabolismUnlike in the general population, thyroid alterations are less frequent than other endocrine disorders in MD1. Thyroid nodules are observed more frequently in these patients, generally with normal thyroid function.
In the case of primary hypothyroidism, the typical symptoms of hormonal hypofunction may increase muscle weakness and exacerbate the symptoms of dystrophy; early diagnosis and hormone replacement therapy are therefore of great importance.
Thyroid function tests (thyroid-stimulating hormone) and physical examination of the thyroids should be performed annually. If thyroid enlargement or nodules are observed in the physical examination or if thyroid stimulating hormone alterations are detected in 2 tests, a thyroid echography should be performed and the patient should be referred to an endocrinologist.81–83
Corticotropic axisPatients with MD1 may also present alterations to the corticotropic axis (corticotropin-releasing hormone/adrenocorticotropic hormone [ACTH]/cortisol) with adrenocortical dysregulation; the frequency of these alterations is not well characterised. The results obtained to date are variable, and the clinical significance is unclear. These patients may present hyperactivity of the corticotropic axis, with increased baseline cortisol and ACTH levels and flattened circadian rhythm of both hormones. The response of cortisol to stimulation with exogenous ACTH varies. Plasma cortisol levels respond adequately in most patients, although there are cases of hyporesponse, which implies reduced epinephrine reserve, and rarely hyperresponse, in patients with greater numbers of CTG repeats. ACTH levels present an exaggerated response to administration of corticotropin-releasing hormone.
Other authors report low baseline cortisol levels, with reduced cortisol response after stimulation with corticotropin-releasing hormone, and higher mean ACTH levels, which indicates a lack of efficacy of this hormone on the adrenal ACTH receptor.
Insulin and metyrapone stimulation tests yield normal results, as does suppression with dexamethasone.84–86
Routine evaluation of the corticotropic axis is not needed; however, in patients presenting clinical signs of hypocortisolism (asthenia, orthostatic hypotension, hyperkalaemia, or hyponatraemia), blood cortisol determination at 08:00 should be requested, with referral to the endocrinology department if the value is below 18μg/dL. Patients with symptoms compatible with hypercortisolism (e.g., central obesity, capillary fragility, facial plethora, reddish striae on the adbomen) should also be referred to endocrinology. Screening for hypercortisolism requires 24-hour urinary free cortisol, salivary cortisol, or dexamethasone suppression test (1mg at 23:00 and cortisol determination at 08:00). If blood cortisol after suppression is greater than 1.8μg/dL, or if elevated urinary or salivary cortisol levels are detected, the patient should be referred to the endocrinology department.
Isolated cases have also been observed of hydroelectrolytic alterations, the cause of which is not well understood. Some patients present hyperkalaemia, which appears to be caused by hyperreninaemic hypoaldosteronism87; other cases are reported of elevated sodium levels with no clinical impact, which may be explained by altered osmoregulation of sodium secondary to deficient vasopressin production and reduced thirst.88
Plasma ion concentrations should be tested annually; if alterations are detected, the test should be repeated and urine sodium and potassium levels should be determined. If electrolyte alterations persist, the patient should be referred to an endocrinologist.
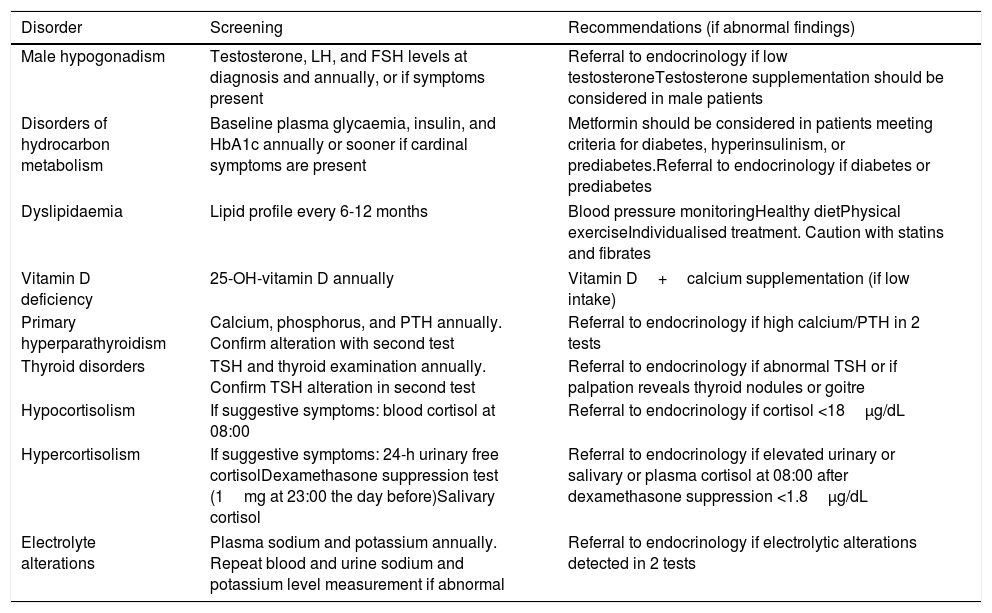
Table 5 shows the recommendations for endocrinology follow-up.
Endocrinology recommendations.
| Disorder | Screening | Recommendations (if abnormal findings) |
|---|---|---|
| Male hypogonadism | Testosterone, LH, and FSH levels at diagnosis and annually, or if symptoms present | Referral to endocrinology if low testosteroneTestosterone supplementation should be considered in male patients |
| Disorders of hydrocarbon metabolism | Baseline plasma glycaemia, insulin, and HbA1c annually or sooner if cardinal symptoms are present | Metformin should be considered in patients meeting criteria for diabetes, hyperinsulinism, or prediabetes.Referral to endocrinology if diabetes or prediabetes |
| Dyslipidaemia | Lipid profile every 6-12 months | Blood pressure monitoringHealthy dietPhysical exerciseIndividualised treatment. Caution with statins and fibrates |
| Vitamin D deficiency | 25-OH-vitamin D annually | Vitamin D+calcium supplementation (if low intake) |
| Primary hyperparathyroidism | Calcium, phosphorus, and PTH annually. Confirm alteration with second test | Referral to endocrinology if high calcium/PTH in 2 tests |
| Thyroid disorders | TSH and thyroid examination annually. Confirm TSH alteration in second test | Referral to endocrinology if abnormal TSH or if palpation reveals thyroid nodules or goitre |
| Hypocortisolism | If suggestive symptoms: blood cortisol at 08:00 | Referral to endocrinology if cortisol <18μg/dL |
| Hypercortisolism | If suggestive symptoms: 24-h urinary free cortisolDexamethasone suppression test (1mg at 23:00 the day before)Salivary cortisol | Referral to endocrinology if elevated urinary or salivary or plasma cortisol at 08:00 after dexamethasone suppression <1.8μg/dL |
| Electrolyte alterations | Plasma sodium and potassium annually. Repeat blood and urine sodium and potassium level measurement if abnormal | Referral to endocrinology if electrolytic alterations detected in 2 tests |
Digestive system involvement is one of the most frequent alterations recorded in MD1, but has not been well studied. Patients and clinicians often give little importance to gastrointestinal alterations. The main cause of these symptoms is the effect of MD1 on smooth muscle. The frequency and intensity of these symptoms are highly variable, and they can be very severe and may represent the initial form of presentation of the disease. A proposed cause is ribonuclear inclusions and muscleblind-like protein 1 (MBNL-1) in smooth muscle nuclei.89 Other hypotheses suggest deficient innervation of smooth muscle, fatty infiltration or fibrosis of the walls of the hollow organs, and even degeneration of smooth muscle. However, no conclusive data supporting these hypotheses have been reported.90–93
Mastication alterationsMastication is impaired as a result of myotonia and weakness of the oral and masticatory muscles (tongue, soft palate, pharynx, masseters, pterygoids). These muscles should be assessed annually. Physical and speech therapy are recommended if any alteration is detected.5
DysphagiaApproximately 55% of patients with myotonic dystrophy present dysphagia, which is associated with a high risk of aspiration pneumonia and malnutrition, and constitutes a significant cause of morbidity and mortality.92
Dysphagia is not related with the degree of skeletal muscle involvement or muscle disease progression time. This symptom is caused by weakness or myotonia of the muscles of mastication, pharyngeal muscles, upper oesophageal sphincter, and oesophagus. These patients present reduced pharyngeal propulsion and increased waste at this level. These mechanisms, combined with the weakness of the respiratory muscles, increase the risk of respiratory infections.93 Patients with myotonic dystrophy and dysphagia usually have greater difficulty swallowing solid foods; this is unusual, as in most neurological diseases dysphagia to liquids or mixed textures is more frequent. A targeted clinical history interview should be carried out for all patients, with emphasis on symptoms suggestive of dysphagia: coughing while swallowing, respiratory infections, and weight loss.79 A simple screening test that may assist in detecting dysphagia is the 10-item Eating Assessment Tool (EAT-10),94 of which a validated Spanish-language version is available. Patients with a positive result should undergo further testing by trained personnel, including the volume-viscosity swallow test, fibre-optic endoscopic evaluation of swallowing, or video fluoroscopy. In the former test, the patient is given different volumes and textures of food, and alarm signs (cough, desaturation, vocal changes) are monitored. This enables identification of safe textures and volumes of food. Fibre-optic endoscopic evaluation is a direct method performed by otorhinolaryngologists to detect penetration or aspiration of different volumes and textures of foods; as it is a direct method, it enables detection of silent aspirations that may go unnoticed in the volume-viscosity swallow test.95 Finally, video fluoroscopy, which is considered the gold standard technique, is a real-time imaging technique used to study swallowing efficiency and safety. However, this technique is costly and is not available at many centres.
Treatment involves adapting foods to the safest texture for each patient. Thickeners should be used if dysphagia affects swallowing of liquids, and mixtures of different textures should be avoided; if swallowing of solid food is affected, soft or blended foods should be given and the foods representing the greatest risk in each patient should be avoided. Guided physiotherapy with a speech therapist may improve this symptom.
It should be noted that dysphagia is associated with a risk of malnutrition; therefore, it is important to monitor patients’ weight in order to detect involuntary weight loss or reduced food intake. We should also monitor biochemical parameters associated with malnutrition: albumin and prealbumin levels. If malnutrition or weight loss are detected, the calorie and protein content of the patient's diet should be increased; if this is not sufficient, nutritional supplements should be added. Some patients will need enteral nutrition, via nasogastric tube if early resolution is expected (< 4-6 weeks), or ostomy sugery (gastrostomy or jejunostomy) if the disorder is expected to persist.
In addition to diet, patients should be prescribed individualised physical exercise to preserve muscle mass or minimise muscle loss as much as possible.
Recommendations regarding dysphagia:
- 1.
Symptoms of dysphagia to solids, liquids, or mixed textures should be monitored at least once per year.
- 2.
The EAT-10 scale should be administered annually.
- 3.
Patients scoring ≥ 3 should also undergo a volume-viscosity swallow test, fibre-optic endoscopic evaluation of swallowing, or video fluoroscopy, depending on the equipment available.
- 4.
Patients should be weighed annually, or more frequently if they present dysphagia.
- 5.
Specific nutritional recommendations are provided for patients with dysphagia (Supplementary Material).
- 6.
In patients presenting malnutrition or involuntary weight loss, food should be enriched, and nutritional supplements should be prescribed in patients presenting > 10% weight loss over a 6-month period or if no changes are observed after implementing the nutritional recommendations.
- 7.
Individualised programmes of physical exercise should be encouraged at all patient consultations.
- 8.
As in other neurological diseases or cases of dysphagia, a nasogastric tube or gastrostomy (depending on disease progression time) are advised in cases of severe dysphagia in which it is not possible to implement specific recommendations.
Oesophageal myotonia, stomach hypotonia, cardia weakness, and poor gastric and oesophageal peristalsis have been reported to cause problems in these patients.
This delayed passage through the oesophagus may be observed with cineradiography or oesophageal manometry; a much simpler, quicker, and less costly technique is a radionuclide oesophageal transit study, which presents excellent correlation with oesophageal manometry.96–99
Gastric emptying is slow, which results in heavy, prolonged digestion and a sensation of abdominal bloating. This alteration, combined with cardia weakness, favours regurgitation and increases the risk of bronchoaspiration. It may be treated with proton pump inhibitors and smooth muscle prokinetic agents.79 A radionuclide gastric emptying study can confirm presence of this alteration.100
GutWeakness and lack of peristalsis of the large and small intestines cause alternating phases of constipation and diarrhoea and abdominal discomfort of varying intensity, and may cause intestinal pseudo-obstruction. Cases of megacolon and volvulus have been described in association with MD1. Radiological studies may reveal segmental dilation and absence of colon haustra due to the reduced peristalsis.100–103
Constipation should be assessed annually. It improves with diets rich in fibre, prokinetic agents, and laxatives.5
Diarrhoea is caused by bacterial overgrowth, and is usually accompanied by swollen abdomen, tympanites, and discomfort. The possibility of nutritional deficiencies, and especially vitamin B12 deficiency, should be considered. Diagnosis is clinical. A test of carbohydrate metabolism, the d-xylose absorption test, exists but is rarely used. Treatment includes diet, probiotics, cholestyramine, and antibiotics (rifaximin at 400mg every 12hours for 7 days each month or ciprofloxacin at 500mg every 12hours for 7 days each month).104
GallbladderPatients with MD1 present elevated incidence of gallstones (25%-50% of cases) due to slow movement of the gallbladder, which favours increased biliary sludge and the formation of gallstones.105
AnusPatients may present severe myotonia of the internal anal sphincter and a mixture of milder myotonia and weakness of the external sphincter, which contributes to constipation. Myotonia may be observed in an electromyography study.106
Genitourinary systemIn addition to the endocrine alterations discussed above, which are associated with hypogonadism, erectile dysfunction, and reduced libido and fertility, patients may present other genitourinary symptoms due to smooth muscle involvement.
For example, women may present inadequate uterine contractions, delayed uterine relaxation, and increased labour duration during childbirth. The risk of postpartum bleeding and retained placenta due to uterine atony is also increased.34
The bladder is usually unaffected in patients with MD1, although the ureters may present focal dilation.34
StomatologyCavities, gingivitis, bacterial plaqueDental alterations are particularly frequent due to hyposalivation and poor oral hygiene, particularly in the posterior dental arch. The oral cavity should be examined annually and good oral hygiene should be emphasised; patients should be given instruction on proper tooth brushing, the use of dental floss and clorhexidine mouthwashes, and avoiding sugary foods, which promote cavities.107,108
Facial dysmorphism, high-arched palate, retrognathia, and prognathismPatients with congenital MD1 may present dysmorphism due to alterations during embryonic development. Orthognathic surgery may be indicated in patients with severe alterations that affect feeding or language.109
Restricted mouth opening, pain, and jaw claudication during masticationThese alterations are caused by temporomandibular joint dysfunction due to morphological changes to the articular disc, with remodelling of the bone and weakness of the masticatory muscles. Imaging studies (functional radiography, MRI) of the temporomandibular joint are used to study these alterations. Mouth splints and physiotherapy may be helpful for patients with pain.110
OphthalmologyCataractsCataracts are very frequent over the course of MD1; these patients develop very peculiar iridescent, subcapsular cataracts. These are present in 90% of patients and are usually diagnosed after the age of 50, although they may appear earlier. In patients with 50-100 CTG trinucleotide repeats, early development of cataracts may be the sole manifestation of the disease. All patients with MD1 should be assessed regularly by an ophthalmologist, at least once every 2 years.
Ophthalmological assessment, including slit-lamp examination, is the typical diagnostic procedure. Cataracts are treated surgically, with removal of the crystalline.111–114
Ocular hypotonyAlmost all patients with MD1 present ocular hypotony, with glaucoma being very rare. This alteration is caused by smooth muscle weakness or ciliary body detachment. It generally causes no symptoms and is typically diagnosed by measuring intraocular pressure, which is usually below 5mmHg. No treatment is required unless functional or structural changes to the eye are detected.115
Palpebral ptosisPalpebral ptosis, caused by weakness of the levator palpebrae superioris muscle, is usually a constant feature of MD1. This symptom is progressive and contributes to these patients’ characteristic facial appearance. Blepharoplasty should be considered in patients with reduced visual field.116
PregnancyFemale patients with MD1 may transmit the disease to their children, who will typically display a more severe form of the disease. Prenatal diagnosis is possible, and severity may be estimated approximately according to the number of CTG trinucleotide repeats.
Women with MD1 more frequently present hydramnios, miscarriage, weakness during labour, retained placenta, and postpartum bleeding.117 A study into pregnancy in patients with MD1 reports premature birth in 19% of cases, urinary infections in 13%, placenta praevia in 9%, and ectopic pregnancy in 4%.118 Given these risks, as well as the possibility of cardiac complications in the mother and the implications of the disease with respect to anaesthesia, these should be considered high-risk pregnancies and special attention should be paid during obstetric follow-up.
AnaesthesiaCertain specific risks should be taken into account when administering anaesthesia. These patients frequently require cataract surgery and abdominal surgery, particularly cholecystectomy, as well as surgical treatment for other complications.21,119
Local anaesthesia is safe, but opiates and sedatives with respiratory suppression should be used with caution, as patients with MD1 are particularly sensitive to these drugs117–120 and may even present paradoxical reactions to muscle relaxants. Spinal anaesthesia is the most advisable technique for surgical delivery.
Such drugs as propofolol or short-acting opiates (e.g., remifentanil) may be used for general anaesthesia.119 In the 24hours following surgery, respiratory function should be monitored by pulse oximetry due to the risk of apnoea21,117,120 and atelectasis secondary to hypoventilation, particularly in patients with existing alterations in forced vital capacity. Heart rate monitoring is also essential in these patients due to the increased risk of cardiac arrhythmia.120 The risk of bronchoaspiration should also be taken into account.
A respiratory assessment should always be performed prior to administration of general anaesthesia.
Patients treated with mexiletine should not receive class I antiarrhythmics due to the risk of conduction block. Depolarising muscle relaxants are contraindicated as they may induce hyperkalaemia. If muscle relaxants are unavoidable, short-acting drugs are recommended; the effect of the drug may be more prolonged than usual. Anticholinesterase inhibitors such as neostigmine may cause severe muscle weakness.
The German Society of Anaesthesiology and Intensive Care Medicine has published a set of guidelines for anaesthesia in patients with MD1; a Spanish-language translation is available on the Orphanet website.119
CancerIncreased incidence of tumours has been reported among patients with MD1.
The first tumours described in these patients are pilomatricomas: benign, often calcified skin tumours arising from hair matrix cells. Numerous articles report tumours of different lineages, including a possible association with basal cell carcinomas. One study analysed data from Swedish and Danish registries of patients with myotonic dystrophy between 1977 and 2008, finding a higher than expected incidence of tumours.121 Compared against the general population, these patients more frequently develop tumours of the endometrium, brain, ovaries, and colon.122 That study did not differentiate between type 1 and type 2 myotonic dystrophy, although it is reasonable to assume that most patients would have had MD1, given the relative prevalence of both diseases. Data collected by the Mayo Clinic between 1993 and 2010 in a cohort of 307 patients with MD1 and MD2 showed a potential increase in the risk of thyroid cancer, choroidal melanoma, and prostate and testicular cancer.123 Another population study performed in Utah found increases in the incidence of endometrial and testicular tumours and non-Hodgkin lymphomas.124–126 These data have been validated in a Spanish cohort study, which found greater incidence of endometrial and ovarian tumours in women and thyroid and brain tumours in men.127
The cause of this apparent increase is not known, although numerous hypotheses have been proposed that involve beta-catenin or downregulation of a family of suppressive microRNAs (200c/141).127 Interestingly, increased incidence of tumours is not reported in other diseases caused by trinucleotide repeat expansions.
It is important to be aware of the increased risk of tumours, as cancer is the third leading cause of death in these patients. The cancers presenting increased risk are mainly low-incidence tumours for which screening programmes in the general population have not been shown to be effective. However, patients should be examined at periodic consultations, with particular clinical suspicion in patients presenting symptoms suggestive of cancer, especially cerebral or abdominal/pelvic symptoms or abnormal uterine bleeding. Thyroid palpation is recommended in endocrine assessments, and an imaging study should be requested if alterations are detected. The possibility of choroidal melanoma should be considered during ophthalmological assessment. Monitoring is more important for cancers that are more frequent in the general population, such as skin tumours.
Special considerations for paediatric patientsMD1 typically manifests during adulthood, although 2 variants, the congenital and childhood-onset forms, affect paediatric patients. Both forms present characteristics differentiating them from the classic adult-onset form, although the diagnostic process is similar: clinical suspicion and subsequent confirmation with a specific genetic study.
Congenital myotonic dystrophyThe congenital form of the disease is the most severe, but also the rarest. Incidence ranges between 2.1 and 5.2 cases per 100000 live births.128,129 In Spain, incidence is estimated at 0.8 cases per 10000 live births.130 The disease is transmitted by the mother in the great majority of cases, with a massive intergenerational expansion in the number of trinucleotide repeats. Children with congenital myotonic dystrophy are typically the index case enabling diagnosis of the mother, who will generally present few symptoms, and other family members.
Manifestations of the disease are present from birth, although obstetric history may reveal reduced foetal mobility before delivery: reduced movement, polyhydramnios, and atrial deformations in echography studies, and caesarian delivery due to lack of progress in labour. Difficulties in delivery increase the risk of perinatal asphyxia. Neonates show marked hypotonia associated with weakness and poor movement. Patients show the characteristic facies, with facial palsy and an inverted V shape of the upper lip. Patients frequently present respiratory problems (over 50% of cases) due to diaphragm and intercostal muscle weakness, pulmonary immaturity (increased risk of premature birth), and failure of cerebral respiratory control. Alterations are also observed in sucking/swallowing due to bulbar palsy.131 Joint contractures are common, especially in the form of clubfoot or pes equinovarus. Unlike patients with adult-onset forms of the disease, paediatric patients rarely present cardiac problems, and no clinical or EP signs of myotonia are observed until later in life; therefore, electromyography is not indicated. Increased incidence of ventriculomegaly and malformations of cerebral cortical development are reported.132
Differential diagnosis should consider congenital myopathies and muscular dystrophies, and congenital myasthenic syndromes.
A neonatal mortality rate of 16%-41% is described for congenital myotonic dystrophy.128,133,134 Neonatal mortality is usually attributed to respiratory problems and withdrawal of support from patients treated with mechanical ventilation for longer than 4 weeks, with little likelihood of recovery in the short/medium term.135 However, this approach is changing in the light of histopathological evidence that congenital myotonic dystrophy is a dysmaturational process, and therefore may improve over time. As a result of this, the duration of mechanical ventilation should not affect the continuity of medical care.128
Patients surviving to the sixth month of life gradually improve, but display clear delays in motor development. Nearly all patients are able to walk independently, and hypotonia and feeding difficulties improve at 3-4 years of age. However, facial weakness continues to be prominent, causing the typical “carp-like mouth.” Myotonia does not appear until adolescence. In contrast to these motor skills, cognitive involvement manifests with variable severity in the early years of life, although these patients have normal intellectual capacity in a minority of cases.136 Mean intelligence quotient is around 70, although intellectual disability may be moderate or severe. This is one of the main problems affecting patients with the congenital form. Children may present autism spectrum traits and other associated behavioural problems.137 Due to their facial appearance, these patients may be treated as though their cognitive capacity is less than it truly is. Language development may be hindered both by weakness of the mouth, palate, and jaw muscles, which affects pronunciation, and by hypoacusia due to recurrent upper respiratory tract infections. Over the years, patients develop distal limb weakness and the other manifestations associated with the adult-onset disease.
Childhood-onset myotonic dystrophyIn the childhood-onset form of the disease, clinical manifestations appear between the first and tenth year of life; pre-, peri-, and postnatal development (first 12 months) is normal. This clinical variant bears little resemblance to the congenital form. The causal mutation may be transmitted by either parent. Motor manifestations may not be as marked initially, with language difficulties and poor academic performance being more apparent. Therefore, a high level of clinical suspicion is needed to diagnose the disease at this age.138 Patients may display behavioural problems, with attention-deficit/hyperactivity disorder being the most frequent; visuospatial function may also be affected, even in patients with a normal intelligence quotient.29 A correlation is reported between the degree of cognitive involvement and the size of the CTG repeat expansion.139
Patients with muscle involvement at onset may display poor coordination and facial and cervical muscle weakness, but do not present the typical appearance of patients with congenital forms.134 Children with this form of the disease may also display distal limb weakness and clinical myotonia, as occurs in older patients. Persistent gastrointestinal problems may present due to smooth muscle involvement. Cardiopathy is rare, usually appearing from the second decade of life. Cataracts, which are typical in adult patients, are not detected in this patient group, although other ophthalmological problems (e.g., refractive errors, reduced visual acuity, and oculomotor alterations) are more frequent.139,140 Over time, these patients develop the same complications as patients with the adult-onset form.
Management of myotonic dystrophy type 1 in paediatric patientsTreatment should follow a multidisciplinary approach, addressing all problems associated with the disease.131 Patients born with congenital myotonic dystrophy should be attended in a neonatal intensive care unit, with particular attention paid to monitoring respiratory function. Some patients may require assisted ventilation, with or without tracheostomy. Limitation of therapeutic effort should be considered in accordance with the individual circumstances of the patient: as mentioned above, prolonged ventilatory support does not necessarily imply poor medium/long-term prognosis. Pulmonology follow-up should be maintained throughout infancy. Respiratory assessment is also necessary in childhood-onset forms due to the possibility of obstructive sleep apnoea or excessive daytime sleepiness. Disorders of sucking and swallowing should be evaluated to avoid aspiration, and feeding by nasogastric tube or gastrostomy should be considered. Given the particular impact of cognitive and behavioural alterations during childhood, a comprehensive psychopedagogical assessment should be performed, which should include assessment of cognitive capacity and executive and visuospatial functions. Clinicians should liaise with the child's school in order to put in place appropriate psychological and educational measures.
Cardiac complications are rare before the second decade of life, although there is a risk of cardiomyopathy and cardiac conduction alterations; therefore, periodic cardiological examinations and annual ECG studies should be performed.141,142
In terms of motor skills, the patient should act in accordance with his/her capacities and physical activity should not be restricted.131 Occupational therapy and targeted physiotherapy programmes may be beneficial. Orofacial problems should be addressed by speech therapists. Orthopaedic treatment (with orthoses or surgery) is indicated in patients with joint deformities.143 Ophthalmological and auditory assessment are advised in order to prevent amblyopia and hypoacusia.
Caution should be exercised when administering anaesthesia, as patients may develop cardiac dysrhythmias or apnoea. Patients should be monitored closely in the hours following the procedure.
Genetic counselling is particularly important with paediatric patients, as young parents may wish to have further children.
Documents for the emergency care of patients with myotonic dytrophy type 1There may be a level of uncertainty among emergency department staff about how to manage medical issues in patients with MD1, whether or not these problems are inherent to the disease itself. With the aim of assisting these patients and emergency physicians treating them, it has been proposed that patients should be given emergency cards to enable fast access to relevant medical information and evidence-based emergency medicine guidelines. This information should be made available to patients and their family members and caregivers and the use of emergency cards should be encouraged.
The French Society for Emergency Medicine conducted a survey to gather data on the expectations of emergency physicians regarding patients with rare diseases.144 Ninety-one percent wished for patients to carry cards showing their personal data and recommendations for treatment of their disease in an emergency. Approximately 75% of respondents wished for an online service enabling them to access clinical practice guidelines for these situations.
In response to the needs identified, new services were created in France to improve the availability of information on rare diseases; this information is available online through the Orphanet Urgences website (“Good practices in emergency departments”) developed by the national medical emergency services and hospital emergency department professionals. This service offers emergency care guidelines and care and emergency cards for some diseases (including MD1), which are available online at no cost.57,145 The level of use is satisfactory, according to data on the number of downloads from the website.146
No similar research has been performed in our setting, although it is reasonable to extrapolate the survey findings to the context of the Spanish healthcare system. In order to address the 2 needs expressed by emergency physicians (information provided by the patient and online information), a “medical alert card” has been prepared for patients with MD1. This document is designed to be carried by patients at all times, and provides information for healthcare professionals who normally would not be familiar with the disease. It includes a brief description of the disease, the contact details of their usual physician, and a QR code linking to the Orphanet emergency guidelines for MD1, and may include updated information on the patient's progression.147
Several organisations have prepared and disseminated guidelines for emergency care of patients with MD1 (the British associations Muscular Dystrophy UK, Myotonic Dystrophy Support Group, and the Scottish Muscle Network, and some hospitals within the British National Health Service).
Despite the risk of the document being lost, damaged, or forgotten and the limitations on the information it may contain, and in addition to the peace of mind it may offer patients, the medical alert card for MD1 essentially aims to enable fast access to information validated by experts through the use of new technology. Unfortunately, it has not yet been possible to quantify the level of use of the document.148
FundingNo funding was received for this study.
Conflicts of interestNone.
The following are the supplementary data to this article:
Please cite this article as: Gutiérrez Gutiérrez G, Díaz-Manera J, Almendrote M, Azriel S, Eulalio Bárcena J, Cabezudo García P, et al. Guía clínica para el diagnóstico y seguimiento de la distrofia miotónica tipo 1, DM1 o enfermedad de Steinert. Neurología. 2020;35:185–206.
This Consensus Guide has obtained the scientific endorsements of the Spanish Society of Neurology (SEN), the Spanish Society of Endocrinology and Nutrition (SEEN) and the Familial Cardiomyopathies Group of the Spanish Society of Cardiology (SEC) and will be published in parallel in the journal Medicina Clínica: http://dx.doi.org/10.1016/j.medcli.2018.10.028, with the consent of the authors and editors.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas