



Today, scientists accept that the central nervous system of an adult possesses considerable morphological and functional flexibility, allowing it to perform structural remodelling processes even after the individual is fully developed and mature. In addition to the vast number of genes participating in the development of memory, different known epigenetic mechanisms are involved in normal and pathological modifications to neurons and therefore also affect the mechanisms of memory development.
DevelopmentThis study entailed a systematic review of biomedical article databases in search of genetic and epigenetic factors that participate in synaptic function and memory.
ConclusionsThe activation of gene expression in response to external stimuli also occurs in differentiated nerve cells. Neural activity induces specific forms of synaptic plasticity that permit the creation and storage of long-term memory. Epigenetic mechanisms play a key role in synaptic modification processes and in the creation and development of memory. Changes in these mechanisms result in the cognitive and memory impairment seen in neurodegenerative diseases (Alzheimer disease, Huntington disease) and in neurodevelopmental disorders (Rett syndrome, fragile X, and schizophrenia). Nevertheless, results obtained from different models are promising and point to potential treatments for some of these diseases.
Hoy en día se acepta que el sistema nervioso central adulto posee una enorme flexibilidad morfofuncional que le permite realizar procesos de remodelación estructural aún después de haber alcanzado su desarrollo y maduración. Además del enorme número de genes que participan en el desarrollo de la memoria, los diferentes mecanismos epigenéticos conocidos también han sido involucrados en procesos de modificación neuronal normal y patológica y, por ende, en los mecanismos de desarrollo de la memoria.
DesarrolloEste trabajo fue llevado a cabo a través de una sistemática revisión de las bases de datos de publicaciones biomédicas sobre los aspectos genéticos y epigenéticos que participan en la función sináptica y la memoria.
ConclusionesLa activación de la expresión génica, en respuesta a estímulos extrínsecos, ocurre también en células nerviosas diferenciadas. La actividad neuronal induce formas específicas de plasticidad sináptica que permiten la formación y almacenamiento de la memoria a largo plazo. Los mecanismos epigenéticos tienen un papel crucial en los procesos de modificación sináptica y en la formación y desarrollo de la memoria. Alteraciones en estos mecanismos producen déficit cognitivo y de memoria en padecimientos neurodegenerativos (enfermedad de Alzheimer y Huntington) así como en trastornos del desarrollo neurológico (síndrome de Rett, X-frágil y esquizofrenia). Los resultados obtenidos en diferentes modelos muestran, sin embargo, un escenario promisorio con tratamientos potenciales para algunos de estos padecimientos.
For some time, it was believed that the adult mammalian brain was an organ unable to maintain structural remodelling processes once its developmental stages were complete. This concept was thought to apply to all structures in the nervous system, and especially to synapses. But these concepts have been revised drastically in recent years, to such an extent that the flexibility of neuronal shape and function in the adult brain is now widely accepted. Neurons and synapses are subject to many types of structural and functional plasticity, and this results in profound changes in inner cerebral structures. These changes adapt to and are mainly generated by neuronal activity patterns, which in turn are stimulated by sensory experience from both internal and external sources.1
Recent discoveries in developmental biology show that activation of gene expression in response to extracellular signals, such as growth factors, also occurs in differentiated postmitotic cells. Transcriptional activation in mature cells may be induced by different extrinsic stimuli, including some of the factors that activate transcription during embryonic and fetal development. More specifically, research has shown that neurons may modify the expression of a group of genes in response to depolarisation stimuli; this finding led to the hypothesis that gene activity may be affected by normal synaptic activity.
The observation that neuronal activity induces both adaptive neuronal changes and changes in gene expression patterns suggests that this sequence of events is the source of specific forms of neuronal, and especially synaptic, plasticity. It has also been shown that the same genes that are stimulated by synaptic activity are at work during brain development; this suggests that plasticity, throughout all stages of brain development, uses similar molecular mechanisms and machinery (Table 1).2
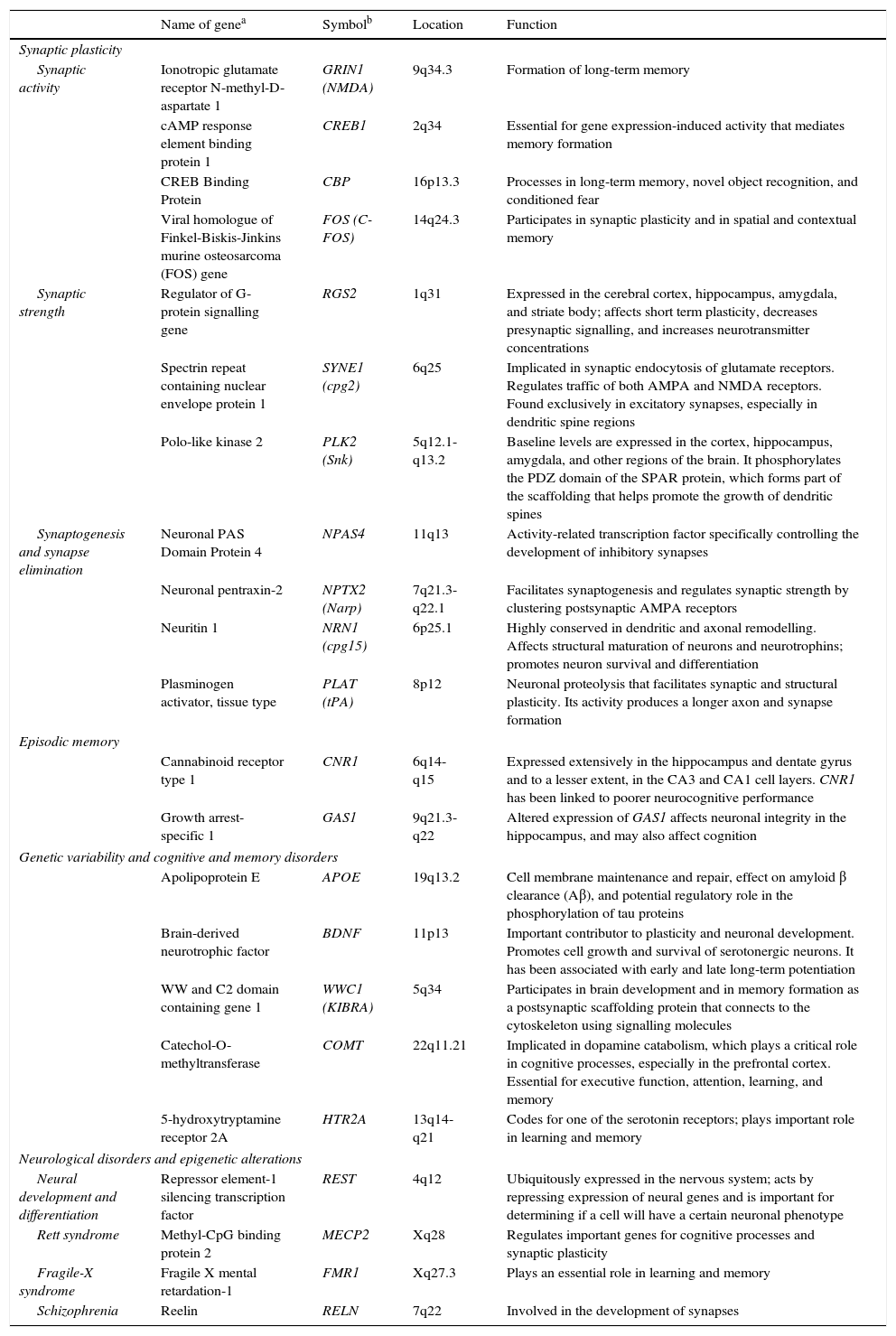
Genes involved in synaptic plasticity, learning, and memory.
| Name of genea | Symbolb | Location | Function | |
|---|---|---|---|---|
| Synaptic plasticity | ||||
| Synaptic activity | Ionotropic glutamate receptor N-methyl-D-aspartate 1 | GRIN1 (NMDA) | 9q34.3 | Formation of long-term memory |
| cAMP response element binding protein 1 | CREB1 | 2q34 | Essential for gene expression-induced activity that mediates memory formation | |
| CREB Binding Protein | CBP | 16p13.3 | Processes in long-term memory, novel object recognition, and conditioned fear | |
| Viral homologue of Finkel-Biskis-Jinkins murine osteosarcoma (FOS) gene | FOS (C-FOS) | 14q24.3 | Participates in synaptic plasticity and in spatial and contextual memory | |
| Synaptic strength | Regulator of G-protein signalling gene | RGS2 | 1q31 | Expressed in the cerebral cortex, hippocampus, amygdala, and striate body; affects short term plasticity, decreases presynaptic signalling, and increases neurotransmitter concentrations |
| Spectrin repeat containing nuclear envelope protein 1 | SYNE1 (cpg2) | 6q25 | Implicated in synaptic endocytosis of glutamate receptors. Regulates traffic of both AMPA and NMDA receptors. Found exclusively in excitatory synapses, especially in dendritic spine regions | |
| Polo-like kinase 2 | PLK2 (Snk) | 5q12.1-q13.2 | Baseline levels are expressed in the cortex, hippocampus, amygdala, and other regions of the brain. It phosphorylates the PDZ domain of the SPAR protein, which forms part of the scaffolding that helps promote the growth of dendritic spines | |
| Synaptogenesis and synapse elimination | Neuronal PAS Domain Protein 4 | NPAS4 | 11q13 | Activity-related transcription factor specifically controlling the development of inhibitory synapses |
| Neuronal pentraxin-2 | NPTX2 (Narp) | 7q21.3-q22.1 | Facilitates synaptogenesis and regulates synaptic strength by clustering postsynaptic AMPA receptors | |
| Neuritin 1 | NRN1 (cpg15) | 6p25.1 | Highly conserved in dendritic and axonal remodelling. Affects structural maturation of neurons and neurotrophins; promotes neuron survival and differentiation | |
| Plasminogen activator, tissue type | PLAT (tPA) | 8p12 | Neuronal proteolysis that facilitates synaptic and structural plasticity. Its activity produces a longer axon and synapse formation | |
| Episodic memory | ||||
| Cannabinoid receptor type 1 | CNR1 | 6q14-q15 | Expressed extensively in the hippocampus and dentate gyrus and to a lesser extent, in the CA3 and CA1 cell layers. CNR1 has been linked to poorer neurocognitive performance | |
| Growth arrest-specific 1 | GAS1 | 9q21.3-q22 | Altered expression of GAS1 affects neuronal integrity in the hippocampus, and may also affect cognition | |
| Genetic variability and cognitive and memory disorders | ||||
| Apolipoprotein E | APOE | 19q13.2 | Cell membrane maintenance and repair, effect on amyloid β clearance (Aβ), and potential regulatory role in the phosphorylation of tau proteins | |
| Brain-derived neurotrophic factor | BDNF | 11p13 | Important contributor to plasticity and neuronal development. Promotes cell growth and survival of serotonergic neurons. It has been associated with early and late long-term potentiation | |
| WW and C2 domain containing gene 1 | WWC1 (KIBRA) | 5q34 | Participates in brain development and in memory formation as a postsynaptic scaffolding protein that connects to the cytoskeleton using signalling molecules | |
| Catechol-O-methyltransferase | COMT | 22q11.21 | Implicated in dopamine catabolism, which plays a critical role in cognitive processes, especially in the prefrontal cortex. Essential for executive function, attention, learning, and memory | |
| 5-hydroxytryptamine receptor 2A | HTR2A | 13q14-q21 | Codes for one of the serotonin receptors; plays important role in learning and memory | |
| Neurological disorders and epigenetic alterations | ||||
| Neural development and differentiation | Repressor element-1 silencing transcription factor | REST | 4q12 | Ubiquitously expressed in the nervous system; acts by repressing expression of neural genes and is important for determining if a cell will have a certain neuronal phenotype |
| Rett syndrome | Methyl-CpG binding protein 2 | MECP2 | Xq28 | Regulates important genes for cognitive processes and synaptic plasticity |
| Fragile-X syndrome | Fragile X mental retardation-1 | FMR1 | Xq27.3 | Plays an essential role in learning and memory |
| Schizophrenia | Reelin | RELN | 7q22 | Involved in the development of synapses |
More recently, scientists demonstrated that gene expression underlying synaptic and neuronal activity is largely regulated by epigenetic mechanisms of the same type as those occurring in embryonic or fetal development. The evidence suggests that epigenetic modifications within the CNS, crucial for behavioural adaptation in the short and long term, are the result of a variety of environmental stimuli. Activation or silencing of these genes, determined by epigenetic mechanisms, seems to constitute an important regulator of synaptic potential and memory.
This study aims to present an updated systematic review of the epigenetic mechanisms related to synaptic function and memory, and of the epigenetic changes involved in memory disorders, whether they are caused by neurodevelopmental anomalies or by neurodegenerative disease.
Epigenetic mechanisms‘Epigenetics’ was coined by Waddington to refer to the array of hereditary processes that regulate gene expression without altering the DNA nucleotide sequence.3 At this date, research has uncovered 3 mechanisms that participate extensively in gene regulation: (1) histone modification, (2) DNA methylation, and (3) non-coding ribonucleic acids (ncRNA). Histone modification is the best understood of these processes, and it is related to numerous neurological diseases.4–6
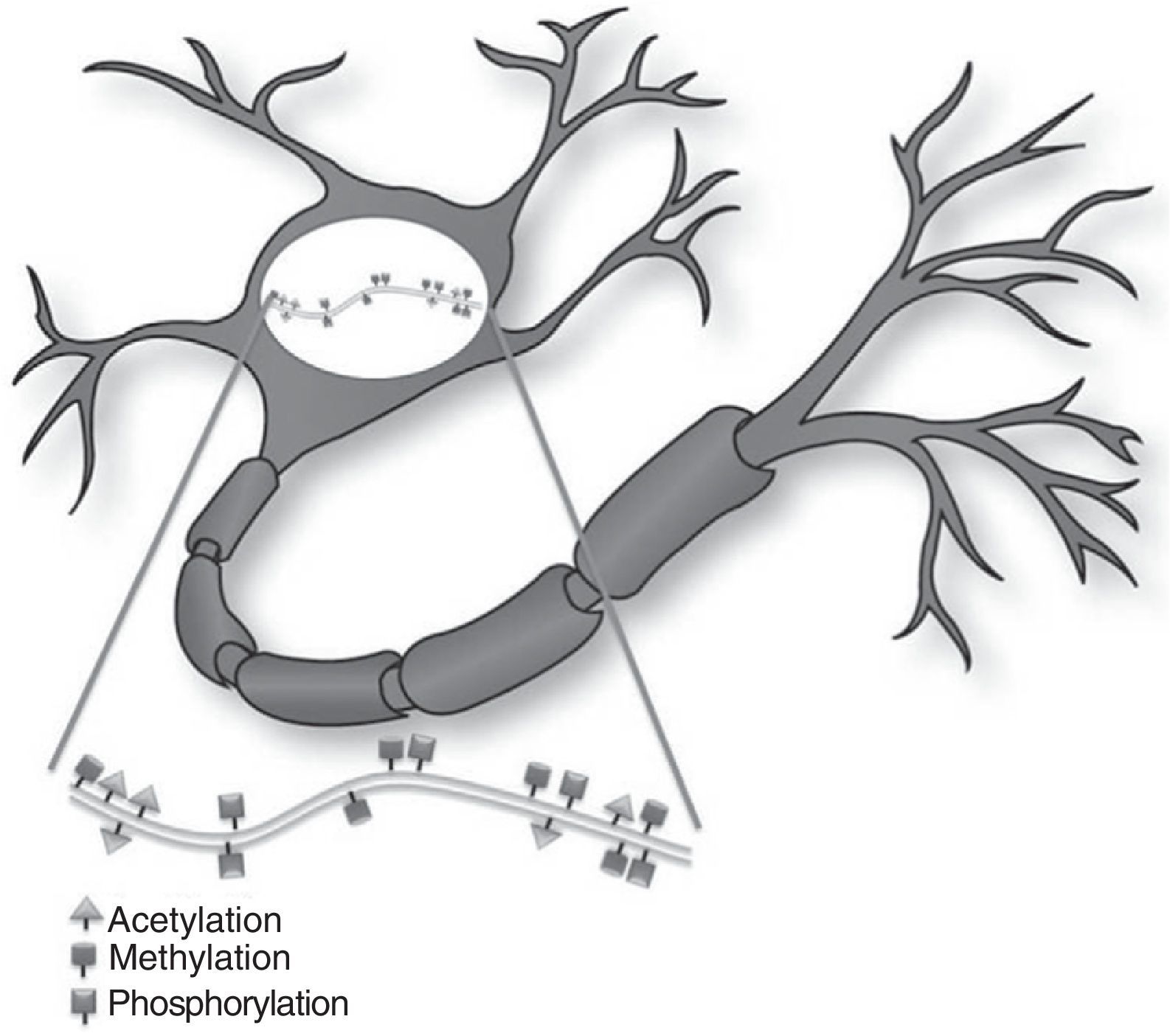
Histone modificationHistones are basic proteins that participate in DNA packaging and in forming nucleosomes. Histone modification is a mechanism that occurs independently or as a result of DNA methylation.6,7 The current hypothesis indicates that histone n-terminal tails, which extend beyond the chromatin structure, participate in a signal integration process that produces a specific pattern of post-translational modifications. This ‘histone code’ directs the activity of numerous transcription machinery factors and cofactors. We now recognise 4 types of post-translational modifications on the tails of histone proteins that participate in epigenetic marking: (1) acetylation, (2) methylation, (3) ubiquitination, and (4) phosphorylation.6,8,9
DNA methylationDNA presents regions of 1000 to 1500 base pairs rich in CpG dinucleotides (CpG islands), which are recognised by DNA-methyltransferase enzymes. During DNA replication, cytosines in the recently synthesised chain are methylated, which serves to maintain memory of the methylated state in the DNA daughter molecule. In general, this modification is stable and inherited as a clonal pattern of methylation. Overmethylation of these regions maintains the condensed form of chromatin and prevents transcription of involved genes.6,8,9
Non-coding RNAThis type of RNA does not code for proteins but its sequences are complementary to encoding DNA or RNA and they prevent translation. This constitutes a type of negative regulation of gene expression at the post-transcriptional level. Non-coding RNA includes small interfering RNA (siRNA), which binds to complementary sequences of mRNA to degrade the molecule and prevent protein translation.6,8,9
Epigenetic mechanisms in synaptic function and memory formationMemory is defined as the brain process enabling long-term storage of information.8 Recent evidence suggests that a wide variety of environmental stimuli produce epigenetic modifications in the CNS that are critical for short- and long-term behaviour modification (Fig. 1).10 As such, epigenetic modifications are active in the creation and maintenance of behavioural memory on multiple levels, and by means of different mechanisms. These studies and others have garnered interest in the therapeutic potential of drugs that are able to improve or change these reactions in cognitive disorders.8,11

The study by Levenson et al.12 showed that formation of long-term memory is also subject to epigenetic marking. In the contextual fear conditioning model, a hippocampus-dependent learning model in which the animal learns to associate a new environment with an aversive stimulus, acetylation of the H3 histone, but not of H4, was found to be significantly increased. The formation of long-term memory in the contextual fear model requires glutamate receptor ionotropic, NMDA (GRIN1), and the MEK-ERK/MAPK signal cascade in the hippocampus; inhibition of either of these elements will impede acetylation of histone H3. These findings indicate that, in the histone code, specific types of memory are associated with specific types of histone modifications.12
Other authors have examined long-term memory formation in transgenic mice with alterations in the functions of CREB-binding proteins (CBP). These transgenic mice are heterozygous for a dominant-negative or truncated form of CBP (CBPDN+/−)105 and they display a significant deficit in different types of long-term memory, such as passive place avoidance, novel object recognition, and conditioned fear.13
Two other independent studies used CBP-deficient mice, but these subjects did not exhibit the serious developmental problems observed in CBPDN+/− mice. In the first of these series, the dominant negative allele of CBP was transformed into an inducible promoter (CBPI-DN+/−).14 Activation of the dominant negative allele after normal development in these animals gave rise to alterations in learning as shown by water maze and novel object recognition tests.14 In the second study, mice heterozygous for CBP (CBP+/−) displayed alterations in contextual and conditioned fear memory, and they also displayed abnormal novel object recognition.15 In both studies, administering an inhibitor of histone deacetylase (HDAC) restored formation of long-term memory. This means that any change to processes that regulate the chromatin structure may affect the formation of long-term memory in vivo.12 HDAC inhibitors did not affect short-term memory in either study.8
Synaptic plasticityChanges in the degree of synaptic activity are widely held to be critical for the formation of long-term memory. There are many studies describing the mechanisms responsible for the induction, expression, and maintenance of synaptic plasticity in different species.16 One intriguing observation is that these mechanisms are similar to those involved in forming long-term memory. Induction of synaptic plasticity may therefore involve epigenetic mechanisms similar to those active in long-term memory formation. One example of this sensory and motor synapse mechanism has been described in the marine mollusk Aplysia as 2 forms of plasticity: long-term facilitation (LTF), referring to lasting improvement of synaptic transmission, and long-term depression (LTD), a lasting decrease in synaptic transmission. Acetylation of histone H4 in the promoter of the CCAAT-enhancer-binding proteins (or C/EBPs) in Aplysia temporarily increased during LTF, but also temporarily decreased during STF. These 2 opposite forms of plasticity therefore induce opposite changes in acetylation of histones in Aplysia.17
Epigenetic changes induced by synaptic plasticity can also be observed in mammalian models. Direct activation of GRIN1 receptors in the hippocampus increases the acetylation of H3 histones, which may be blocked by the inhibition of the MEK-ERK/MAPK cascade. In the same way, the activation of dopaminergic, cholinergic, and glutaminergic pathways in the hippocampus induce phosphorylation of H3 histones that depend on the increase in ERK.12 These results suggest that inducing synaptic plasticity in mammals results in an ERK-dependent increase in histone acetylation and phosphorylation in the hippocampus. This process is similar to that observed in Aplysia. These studies indicate that the epigenetic state of the genome has a long-term effect on the induction of synaptic plasticity in mammals.8,12
Neurological diseases and epigenetic changesAn increasing body of evidence points to epigenetic mechanisms as a cause of human cognitive dysfunction. Neurodegenerative diseases and neurodevelopmental disorders may be attributed, at least partially, to mechanisms underlying epigenetic marking of the genome. The next section presents the findings related to some of the more interesting examples of neurological disorders.
Neurodegenerative diseaseSome neurodegenerative diseases appear to be related to altered epigenetic mechanisms. These include Alzheimer disease (AD) and Huntington disease (HD), which have been thoroughly studied (Table 2).
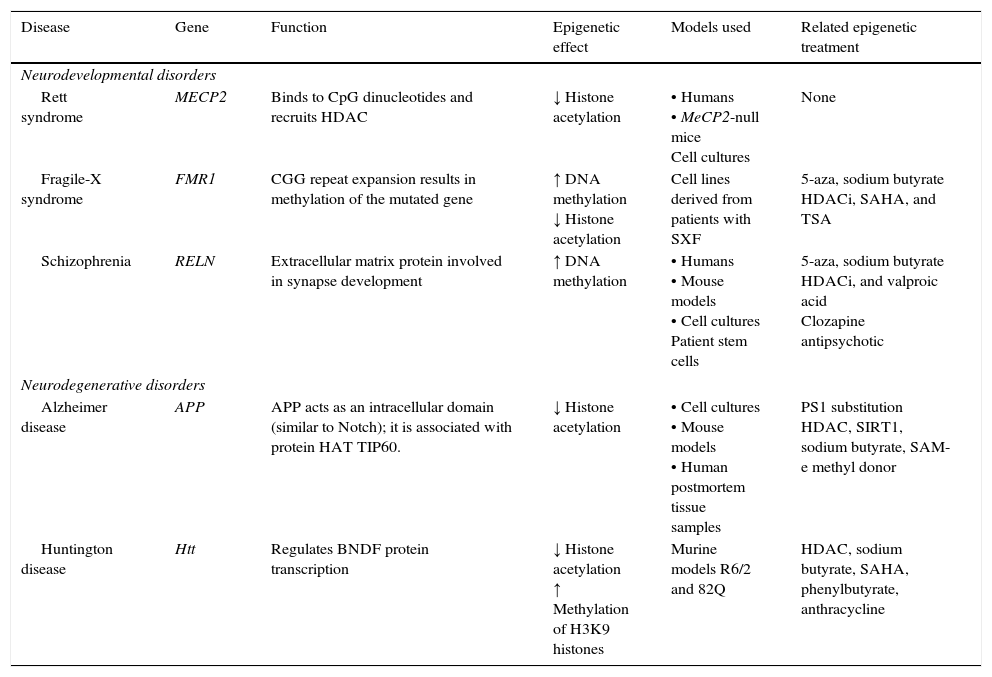
Epigenetic mechanisms and therapies in neurodegeneration and neurodevelopmental disorders.
| Disease | Gene | Function | Epigenetic effect | Models used | Related epigenetic treatment |
|---|---|---|---|---|---|
| Neurodevelopmental disorders | |||||
| Rett syndrome | MECP2 | Binds to CpG dinucleotides and recruits HDAC | ↓ Histone acetylation | • Humans • MeCP2-null mice Cell cultures | None |
| Fragile-X syndrome | FMR1 | CGG repeat expansion results in methylation of the mutated gene | ↑ DNA methylation ↓ Histone acetylation | Cell lines derived from patients with SXF | 5-aza, sodium butyrate HDACi, SAHA, and TSA |
| Schizophrenia | RELN | Extracellular matrix protein involved in synapse development | ↑ DNA methylation | • Humans • Mouse models • Cell cultures Patient stem cells | 5-aza, sodium butyrate HDACi, and valproic acid Clozapine antipsychotic |
| Neurodegenerative disorders | |||||
| Alzheimer disease | APP | APP acts as an intracellular domain (similar to Notch); it is associated with protein HAT TIP60. | ↓ Histone acetylation | • Cell cultures • Mouse models • Human postmortem tissue samples | PS1 substitution HDAC, SIRT1, sodium butyrate, SAM-e methyl donor |
| Huntington disease | Htt | Regulates BNDF protein transcription | ↓ Histone acetylation ↑ Methylation of H3K9 histones | Murine models R6/2 and 82Q | HDAC, sodium butyrate, SAHA, phenylbutyrate, anthracycline |
APP, amyloid precursor protein; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; HAT, histone acetyltransferase; Htt, huntingtin; MECP2, methyl-CpG-binding protein 2; PS1, presenilin; SAHA, suberoylanilide hydroxamic acid; SIRT1, silent mating type information regulation homolog 1; TSA, trichostatin A; 5-aza, 5-aza-2′-deoxycytidine. Modified from: Levenson and Sweatt,8 Scarpa et al.24
AD is the most common cause of primary degenerative dementia. It is characterised by cognitive changes, dementia, and the accumulation of plaques formed by β-amyloid and neurofibrillary tangles in different areas of the brain. It has a prevalence of some 35 million people worldwide.18,19
Recent evidence shows that histone acetylation and DNA methylation contribute to AD aetiology. Amyloid plaques are formed by the deposition of amyloid-β peptides produced during β and γ secretase cleavage of the amyloid precursor protein (APP). Interestingly enough, APP processing results not only in the extracellular amyloid-β fragment, but also in an intracellular fragment named APP intracellular domain (AICD). This fragment interacts in vitro with HAT-TIP60 such that they form a transcriptional activator. These findings suggest that AD is associated with an increase in histone acetylation.18 This theory has been supported by results in neuron cultures, which show that mutations in the presenilin1 gene (PS1, gene coding for a member of the γ-secretase complex) inhibit degradation, via the proteosome pathway, of the HAT domain of CBP. This increases CREB-mediated gene expression.20
A recent in vivo study found that overexpression of the HDAC SIRT1 conferred protection against neurodegenerative disease in a murine model of AD. However, it is not known whether SIRT1 acts by epigenetic mechanisms, through other genes, or using both factors.21 Other studies have also demonstrated decreased histone acetylation, an effect that is causally related to AD.22
Lastly, DNA methylation is another mechanism involved in AD aetiology, with hypomethylation being the most widely reported phenomenon. In cell cultures, hypomethylation of the promoter region of PS1 has increased expression of presenilin and promoted formation of β-amyloid plaques.23–26 Some posit that this effect could be reversed by applying a methyl donor, such as S-adenosyl methionine (SAM), to rescue methylation and decrease the expression of presenilin. That process would reduce formation of β-amyloid plaques. These studies suggest that methyl donors and/or the drugs that act on methyl metabolism are potential therapeutic agents for treating AD.25–27
Huntington diseaseThis rare neurodegenerative disease has an autosomal dominant inheritance pattern and an incidence of 3 to 7 cases per 100000 population in the United States. It is characterised by cognitive impairment and memory loss, psychiatric changes, and uncoordinated movement. HD results from a mutation in the HTT gene consisting of an increase in the number of CAG repeats in the 5′ terminal of the gene, which increases the polyglutamines in the N-terminal region of the huntingtin protein.28
Multiple studies have suggested that histone acetylation and methylation are altered in HD. Mouse models in which the activity of CBP has been compromised demonstrate reduced chromatin acetylation, decreased LTF in the hippocampus, and long-term memory deficit. Loss of the CBP function has also been associated with degeneration of the striate body in mouse models for HD. Furthermore, huntingtin, the product expressed by the mutated HTT gene, interacts directly with the acetyltransferase domains of CBP, resulting in decreased activity by that enzyme.29
Another mechanism that has been observed in cell cultures involves the binding of polyglutamines to histone acetyltransferase (HAT) proteins, to CBP, and to the p300/CBP associated factor; this reduces HAT activity and the level of acetylation of histones H3 and H4. These results show that the decrease in CBP levels, and alterations in CBP/CREB-dependent transcription processes, are associated with deficient long-term memory in HdhQ7/Q111 mutant mice.29
In recent studies carried out in a drosophilia model with polyglutamine expansion, HDAC activity has been decreased through directed mutations or pharmacological inhibition, which reduces the harmful effects induced by polyglutamine.29 The list of currently known HDAC inhibitors includes sodium butyrate, suberoylanilide hydroxamic acid (SAHA), phenylbutyrate, and HDAC inhibitor 4b. These molecules decrease motor deficit and neuronal atrophy in mouse models of HD (Fig. 2).30–34 Researchers have observed an interesting reversal of the hypoacetylated state of cytoplasmic α-tubulin mediated by HDAC inhibitors.20,29 In another study, striate cells from mice with or without HD were treated with tubacin, the selective inhibitor of HDAC6, resulting in increased acetylation of α-tubulin and reversal of some signs of the disease.35

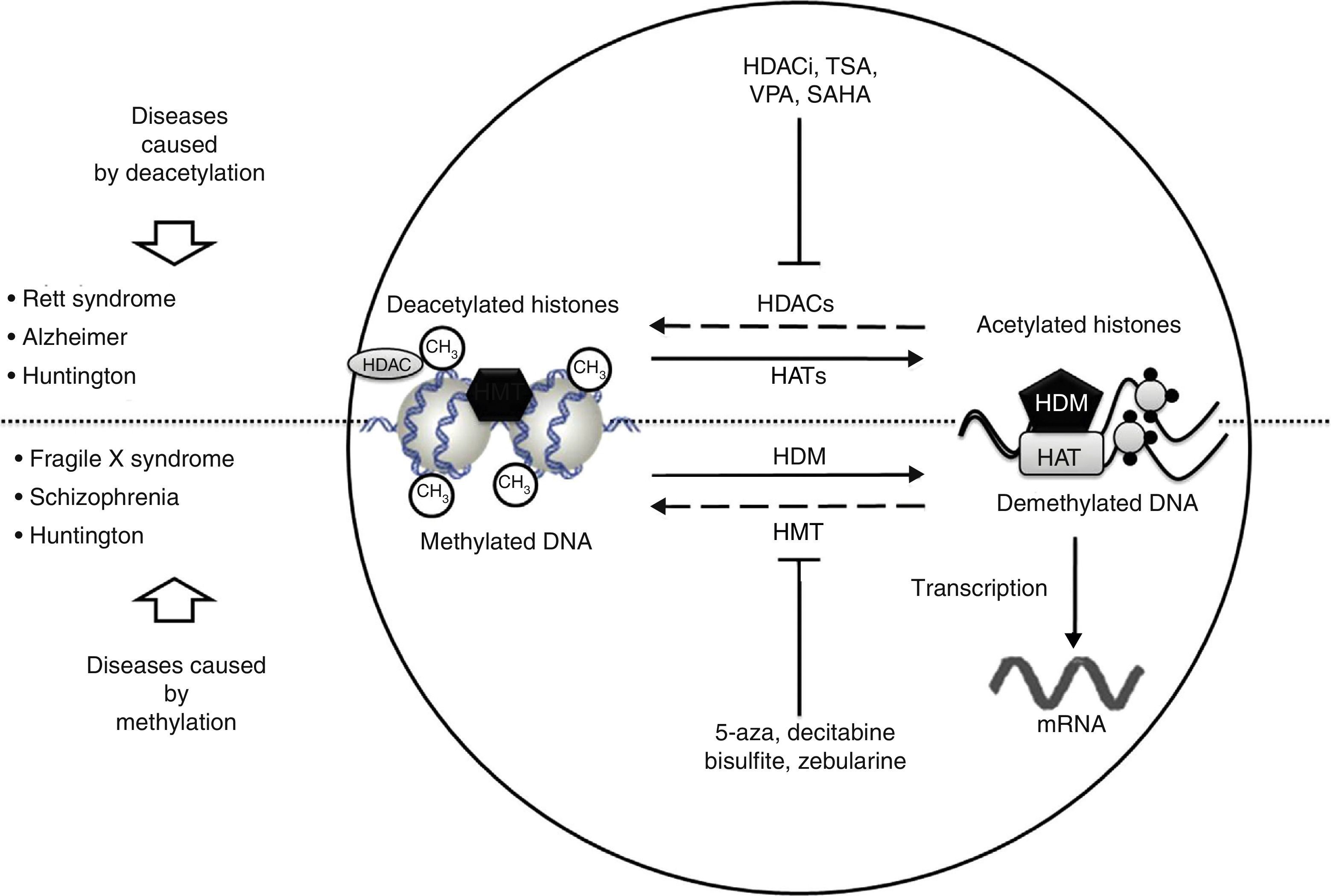
Neurological disorders and epigenetic alterations. Under normal conditions, HAT enzymes acetylate histone tails to permit correct DNA transcription. HDAC enzymes, in contrast, deacetylate histones, and this process can give rise to Rett syndrome, Alzheimer disease, and Huntington disease. Substances including HDACi, TSA, VPA, and SAHA inhibit HDAC enzymes and are used to treat these entities. On the other hand, HDM enzymes demethylate DNA under normal conditions, which permits correct transcription. HMT enzymes methylate DNA, which results in fragile X syndrome, schizophrenia, and Huntington disease. Substances including 5-aza, decitabine, bisulfite, and zebularine inhibit HMT enzymes and are used to treat these entities. HAT, histone acetyltransferases; HDAC, histone deacetylases; HDACi, histone deacetylase inhibitor; HDM, histone demethylases; HMT, histone methyltransferases; SAHA, suberoylanilide hydroxamic acid; TSA, tricostatin A; VPA, valproic acid; 5-aza: 5-aza-2′-deoxycytidine.
Acetylation as well as methylation is altered in mouse models of HD, which display an increase in protein dimethyl-H3K9; this protein participates in transcriptional repression processes.25,27,36
Neurodevelopmental disordersA substantial amount of evidence indicates that epigenetic alterations are involved in the aetiology of several neurodevelopmental disorders, including Rett syndrome (RS), fragile-X syndrome, and schizophrenia (Table 2).
Rett syndromeIn the United States, RS affects between 1 in 10000 and 1 in 15000 population. The disease manifests in early infancy as altered neurological development that causes learning and memory deficits. It is characterised by microcephaly, stereotyped movements, altered motor coordination, convulsions, language and learning difficulties, and mild to severe cognitive impairment. Other clinical manifestations include ataxia, spasticity, and autonomic changes.
The epigenetic mechanism related to RS is methylation of DNA that prevents transcription of the MECP2 gene.37 MECP2 protein is a member of the family of methyl binding transcriptional repressors, and it plays a dual role in gene silencing and transcriptional activation.37 In addition to regulating DNA methylation, MECP2 participates in histone acetylation and methylation (Fig. 2).
Results from studies carried out in mouse models with MECP2 deletions in specific regions of the brain resemble the phenotype exhibited by patients with RS.38 A MECP2-deficient mouse model yielded smaller, lighter brains with smaller neurons. Furthermore, mice exhibited complete absence of exploring activity,39,40 cognitive deficit, and reduced synaptic plasticity.41
Cognitive changes may be reversed by overexpression of MECP2; this is consistent with the effect of MECP2 protein deficiency.27,42,43 Phosphorylation of MECP2 also permits dendritic growth and maturation of dendritic spines.27,44 Researchers working with mouse models have observed that overexpression of MECP2 induces LTF in the hippocampus and improves formation of long-term memory. This demonstrates that MECP2 modulates induction of synaptic plasticity and memory formation.45
Fragile X syndromeFXS is the most common hereditary cause of intellectual disability linked to the X chromosome. The syndrome affects approximately one in 4000 men and one in 8000 women. It is associated with cytogenetic expression of the fragile site Xq27.3 (FRAXA) and it is transmitted by means of an unusual X-linked inheritance mechanism. In addition to an array of dysmorphic characteristics, patients with FXS present cognitive impairment and learning and memory disabilities. The mutation causing FXS is an amplification of CGG trinucleotides in the non-translatable 5′ region of the FMR1 gene.19 It is now understood that DNA methylation, as well as histone acetylation and methylation, are involved in the molecular mechanisms of this disease. Repeats of the CGG trinucleotide result in increased DNA methylation in the promoter of the FMR1 gene, which prevents gene transcription and halts production of mRNA and the FMR1 protein (FMRP).46–48 The FMRP, which is absent in patients with FXS, binds to mRNA and participates in the formation of polyribosomes.46
Recent studies in mouse models of FXS show that translation regulation by FMRP is essential for learning and memory, both in adult neural stem cells and in young neurons. Presence of a low number of new neurons with defective maturation may contribute to the cognitive deficiency seen in FXS.49
Lymphoblastoid cells in patients with FXS are treated with demethylating agents such as 5-aza-2-deoxycitidine, which effectively reverts hypermethylation of the FMR1 promoter and reestablishes levels of mRNA.50 Interestingly enough, reestablished mRNA expression may exceed the level of reference when 5-aza-2-deoxycitidine is combined with HDAC inhibitors, such as 4-phenylbutyrate, sodium butyrate, and TSA.51 This suggests that the mechanism causing FXS is a combined effect of DNA deacetylation and hypermethylation; conversely, the synergic effect of histone hyperacetylation and DNA demethylation may provide a treatment approach for patients with FXS (Fig. 2). Two additional studies report that treatment with 5-aza-2-deoxycitidine in cell lines from FXS patients shifts the epigenetic pattern of histone methylation.52 Other studies using chromatin immunoprecipitation in regions of the FMR1 gene promoter have shown that treatment with 5-aza-2-deoxycitidine decreases methylation of histone H3K9, which participates in transcriptional silencing.25,53
SchizophreniaSchizophrenia is a relatively common mental illness, with a prevalence of 1:100 adults in the United States. It is characterised by 2 types of psychotic symptoms. Positive symptoms include disordered thoughts, illusions, hallucinations, and deliriums. Negative symptoms include social isolation, loss of motivation, and apathy; they reflect a loss of behavioural abilities. Patients also present cognitive impairment, manifesting as loss of working memory and conceptual disorganisation.27
The causes of schizophrenia are not well understood, but they probably result from complex interactions between genetic predisposition and environmental conditions during prenatal and postnatal development. Evidence suggests that epigenetic mechanisms are involved, especially methylation of histones and DNA. The brain tissue of patients diagnosed with schizophrenia shows markedly reduced mRNA levels of reelin (extracellular matrix protein involved in neuronal migration; product of RELN). This decrease correlates with increased levels of DNA methyltransferase 1 protein (DNMT1). According to this finding, DNA hypermethylation in the promoter region of the RELN gene may be responsible for the lower levels of RELN gene in schizophrenic patients.54 The RELN gene promoter has been shown to contain several specific sites for methylation, and inhibition of the activity of HDAC and NMT increases RELN expression. These data point to the importance of epigenetic mechanisms in the expression of reelin.25,45
Later studies have shown increased acetylation of lysines in histone 3 (H3K9 or K14) in the RELN gene promoter in murine brains following treatment with valproic acid in association with clozapine or sulpiride.55,56 On the other hand, studies of neuronal cells, both in vivo and in vitro, show that methylation of the RELN gene suppresses its expression. Treatment with TSA and valproic acid increased DNA methylation in the RELN gene promoter; in mice heterozygous for RELN, valproic acid injections have increased expression of the RELN gene. These findings suggest that an interaction between DNA demethylation and histone acetylation may activate expression of the RELN gene.54
ConclusionsEpigenetic mechanisms such as covalent modification of DNA and post-translational changes in histones have been recognised as the necessary regulators of synaptic physiology and memory. Lastly, epigenetic mechanisms play a central role in brain function, and therefore any changes may lead to abnormal neurological development or neurodegenerative processes. Studies carried out in pathological mouse models have contributed the larger part of our current knowledge in this area, and results are promising; they point to an array of potential treatments, such as HDAC inhibitors, for certain neurological diseases. Nevertheless, and despite the growing body of scientific evidence highlighting the role of epigenetic mechanisms in synaptic modification, we still find an enormous quantity of unresolved questions about these mechanisms and how they control long-term memory consolidation and development.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Rosales-Reynoso MA, Ochoa-Hernández AB, Juárez-Vázquez CI, Barros-Núñez P. Mecanismos epigenéticos en el desarrollo de la memoria y su implicación en algunas enfermedades neurológicas. Neurología. 2016;31:628–638.
