



Autoimmune myopathies are rare, with a prevalence of 9-14 cases per 100 000 population.1 Immune-mediated necrotising myopathy (IMNM) is characterised by proximal muscle weakness, elevated creatine kinase (CK) levels, muscle fibre necrosis with signs of regeneration, and minimal or no inflammatory infiltration in muscle biopsy.2 Development of IMNM has been associated with statin use,3 certain connective tissue disorders, and HIV infection.2,4 According to published series,5 some patients test positive for anti-HMGCR (33%) or anti-SRP antibodies (24%), although up to 20%-30% of patients are seronegative.6 We present a case of immune-mediated necrotising myopathy following treatment with adalimumab in a patient with HLA-B27 ankylosing spondylitis.
Our patient was a 55-year-old man with history of psoriasis and HLA-B27 ankylosing spondylitis of 20 years’ progression, who had been under treatment with adalimumab for the previous 2 years. He consulted due to predominantly proximal muscle pain in the limbs, starting 6 months after onset of adalimumab treatment. The patient reported greater difficulty walking and climbing up and down stairs, and loss of muscle mass in the arms and legs. The neurological examination revealed bilateral paresis (Medical Research Council grade 4/5) in the psoas muscle and quadriceps, and muscle atrophy in the deltoid and quadriceps muscles. We detected no fasciculations or any other spontaneous muscle activity, and the patient did not present scapular winging. Deep tendon reflexes were present and gait was normal.
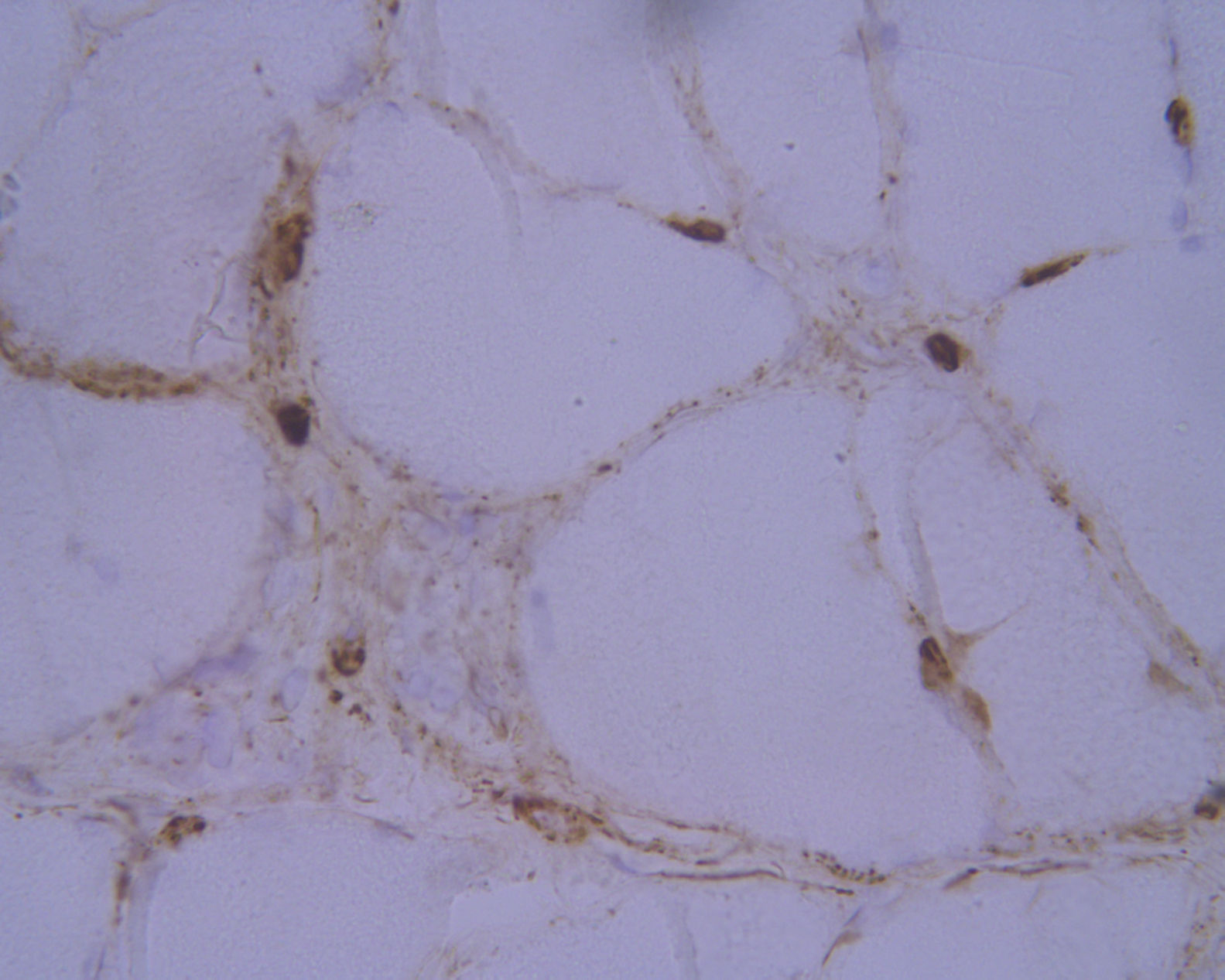
Laboratory analysis revealed elevated CK levels (385 IU/L) at 7 months after treatment onset; results from a complete blood count, erythrocyte sedimentation rate, aldolase level determination, liver profile, kidney profile, and serology testing were all within normal ranges. Due to suspicion of myopathy, we performed an MRI scan of the lower limbs, which showed no alterations, and an electromyography study, finding no signs of myopathy. CK levels were determined periodically, with a maximum level of 598 IU/L. A biopsy of the quadriceps muscle revealed signs compatible with IMNM (Figs. 1 and 2). Tests for antinuclear, anti-ENA, anti-HMGCR, and anti-SRP antibodies and a myositis autoantibody panel yielded negative results. A chest-abdomen-pelvis CT scan ruled out underlying neoplasia.
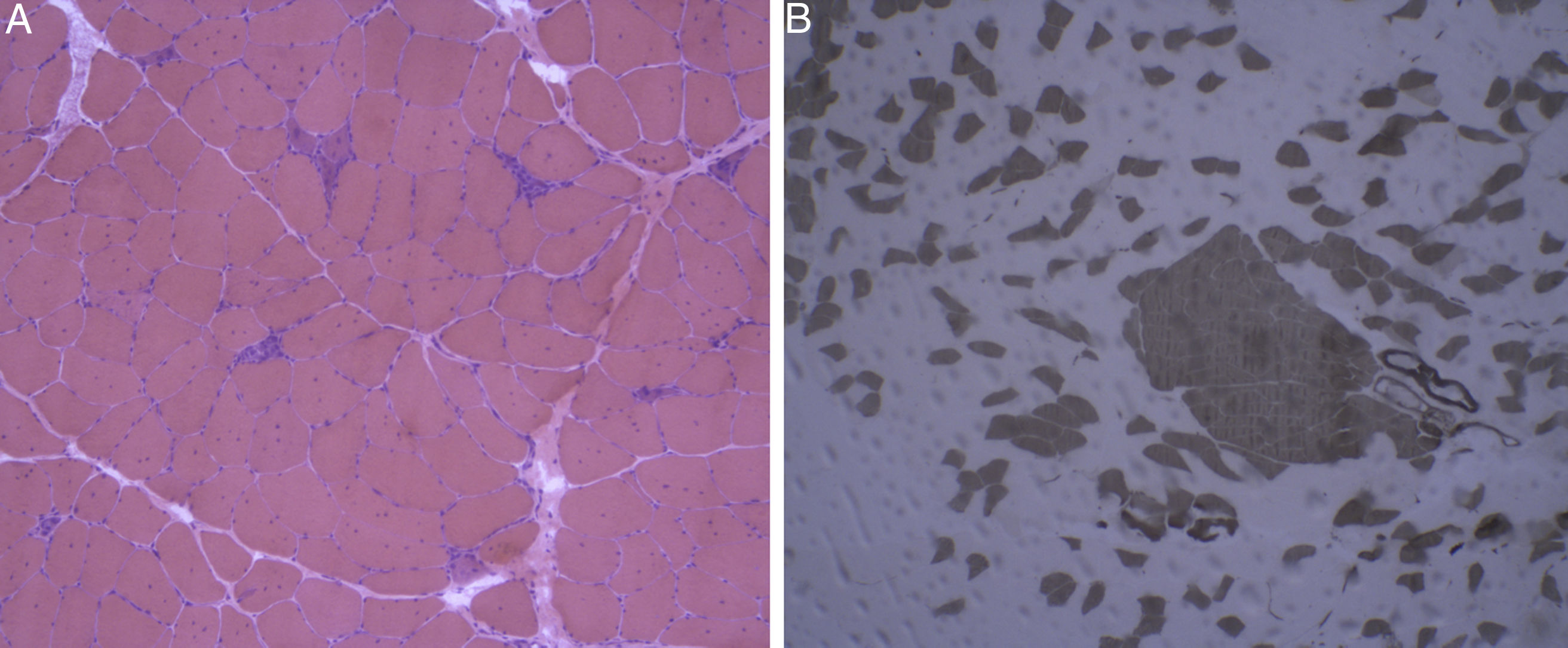
 Haematoxylin-eosin staining: skeletal muscle with normal architecture, without thickening of endomysial connective tissue or adipose tissue infiltration. The image shows fibre regeneration, as well as fibre necrosis, with signs of myophagocytosis. B) ATPase pH 9.4 stain: rearrangement by fibre type, with groups of type 1 and type 2 fibres, a finding suggestive of reinnervation.")
Haematoxylin-eosin staining and immunohistochemical study of a muscle specimen. A) Haematoxylin-eosin staining: skeletal muscle with normal architecture, without thickening of endomysial connective tissue or adipose tissue infiltration. The image shows fibre regeneration, as well as fibre necrosis, with signs of myophagocytosis. B) ATPase pH 9.4 stain: rearrangement by fibre type, with groups of type 1 and type 2 fibres, a finding suggestive of reinnervation.

In view of the anatomical pathology findings, we suspended adalimumab and started corticosteroid therapy, initially with intravenous methylprednisolone at 500 mg for 3 days, followed by oral prednisone in decreasing doses until reaching a maintenance dose of 20 mg/day. At 6 months, CK levels had decreased to 120-130 IU/L and motor deficits had improved slightly, although muscle pain persisted. Azathioprine 50 mg/12 h slightly improved pain.
Tumour necrosis factor-α (TNF-α) inhibitors are increasingly used for the treatment of autoimmune diseases. Paradoxically, due to their immunomodulatory effects, cases have been reported of immune disorders associated with use of TNF-α inhibitors, including vasculitis, lupus-like syndrome, and interstitial lung disease.7 Case series have been published of patients receiving treatment with TNF-α inhibitors and presenting onset of such immune-mediated myopathies as myositis and dermatomyositis.7,8 Despite this potential association, no cases have previously been described of IMNM associated with adalimumab use. In our patient, the short time between onset of treatment with adalimumab and onset of symptoms and the improvement in CK levels and muscle pain after adalimumab discontinuation suggest that this drug played a role in the development of myopathy.
The recommended treatment for IMNM is induction therapy with oral or intravenous corticosteroids; an immunosuppressant may be added at onset of corticosteroid therapy or during the following month, depending on symptom severity and initial response to treatment. Acceptable response has been reported for such immunosuppressants as methotrexate, azathioprine, ciclosporin, and mycophenolate mofetil.9 Combination therapy with several immunosuppressants may be necessary in 50% of cases; intravenous immunoglobulins or rituximab constitute an alternative for refractory cases, or may even be administered during induction therapy.9 In conclusion, TNF-α inhibitors have been associated with the development of immune-mediated myopathy; identification of the causal factor and early discontinuation of the drug are essential for disease prognosis.
Please cite this article as: Chavarría-Miranda A, Hernández Lain A, Toldos González O, Pedraza Hueso MI. Miopatía necrosante inmunomediada tras tratamiento con adalimumab en paciente con espondilitis anquilosante HLA-B27. Neurología. 2021;36:631–632.
