



Treatment-induced diabetic neuropathy (TIDN), previously known as insulin neuritis, is a type of acute neuritis affecting diabetic patients after abrupt re-establishment of metabolic function. The condition was first described in 1933 by Caravati,1 in a diabetic patient who developed symptoms of neuritis following rapid glycaemic control with insulin; symptoms resolved with insulin withdrawal. Few cases have been described to date; TIDN may be underdetected and is often misdiagnosed for other more frequent neuropathies in these patients. We present the case of a patient with diabetes who was admitted with acute neuropathy; symptoms were not initially identified as TIDN but were subsequently controlled.
The patient was a 25-year-old man with a family history of type 1 diabetes mellitus (grandfather and maternal uncle), who developed diabetic ketoacidosis in 2011. He was treated with insulin and informed about the disease and how to manage it. Follow-up was irregular over the first 2 years, as the patient did not attend follow-up appointments. He was admitted to the endocrinology department in 2014; he had discontinued insulin therapy for 3 months, displaying a glucose level >500mg/dL, glycosuria, anorexia, postprandial fullness, and weight loss (BMI 15.5, indicating severe malnutrition). He was oriented and displayed no alterations in level of consciousness. The patient had ketonuria and an HbA1c level of 15.2%. Following rigorous metabolic correction, he was referred to the outpatient endocrinology department. Six weeks later, the patient began to experience pain in the knees and in the scapular and lumbar paraspinal muscles. Over a period of 7-15 days, the pain became burning, and stabbing in the proximal area of the lower limbs (“like having the flesh torn from my bones,” as described by the patient); the pain was associated with dysaesthesias in the soles of the feet. The patient was readmitted due to intense pain (VAS: 8-9). The patient was extremely underweight and in a poor mood, but his level of consciousness, cortical function, and cranial nerve function were normal. Strength was preserved; all reflexes were hypoactive. Regarding superficial sensation, the patient showed hyperaesthesia and allodynia in the paraspinal muscles and in all 4 limbs. Deep sensation, cerebellar function, and gait were normal. The HbA1c level was 9%. High doses of NSAIDs, tramadol, and duloxetine achieved partial relief. The endocrinology department initially diagnosed radiating lower back pain, bone and muscle pain, and reactive depression. A lumbar MRI scan revealed diffuse degenerative changes and disc bulging affecting the entire lumbar segment. During admission, he displayed sustained tachycardia (101-117bpm) and diastolic hypertension (86-92mmHg), with normal systolic blood pressure (105-120mmHg). EMG indicated demyelinating sensorimotor neuropathy, which did not meet the neurophysiological criteria for chronic inflammatory demyelinating polyneuropathy, and no signs of denervation activity (Table 1). A CSF analysis revealed high protein levels (89mg/dL). A complete blood count ruled out systemic, liver, kidney, or thyroid diseases, and vitamin deficiencies (vitamins A, E, D, B1, B6, B9, and B12). The autoimmunity and serology tests for Borrelia, Mycoplasma, CMV, VZV, EBV, HSV, HIV, HBV, and HCV yielded negative results. The levels of thyroid hormones, ACTH, cortisol, iPTH, selenium, zinc, and copper were also within normal ranges. The urine albumin/creatinine ratio was normal. The patient was diagnosed with multifocal demyelinating polyneuropathy associated with poor diabetes control. A follow-up EMG was scheduled at 7 months; the patient refused. Pain progressively improved and the patient was discharged; 6 months later, he was nearly asymptomatic. During that period, the patient gained little weight (3-4kg) and blood glucose levels were partially controlled. As TIDN was not suspected, autonomic function was not assessed; autonomic symptoms were overshadowed by the severe, disabling pain that led to the consultation with the neurology department.
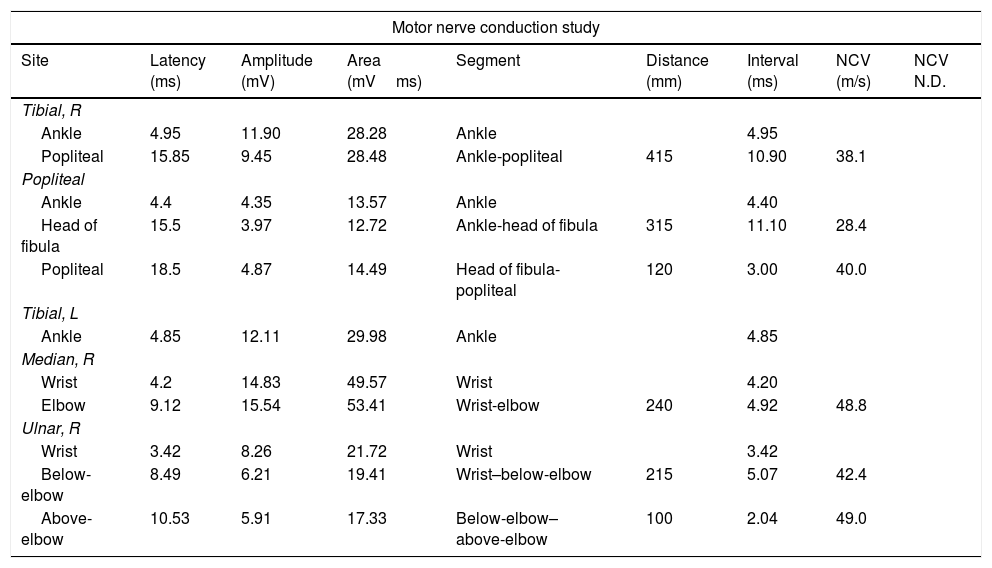
Motor nerve conduction study of the patient revealing slowed conduction velocity in certain motor nerves of the lower limbs (tibial: 38.1m/s; distal peroneal: 28.4m/s) and in the right ulnar nerve (42m/s below-elbow–wrist; normal amplitude).
| Motor nerve conduction study | ||||||||
|---|---|---|---|---|---|---|---|---|
| Site | Latency (ms) | Amplitude (mV) | Area (mVms) | Segment | Distance (mm) | Interval (ms) | NCV (m/s) | NCV N.D. |
| Tibial, R | ||||||||
| Ankle | 4.95 | 11.90 | 28.28 | Ankle | 4.95 | |||
| Popliteal | 15.85 | 9.45 | 28.48 | Ankle-popliteal | 415 | 10.90 | 38.1 | |
| Popliteal | ||||||||
| Ankle | 4.4 | 4.35 | 13.57 | Ankle | 4.40 | |||
| Head of fibula | 15.5 | 3.97 | 12.72 | Ankle-head of fibula | 315 | 11.10 | 28.4 | |
| Popliteal | 18.5 | 4.87 | 14.49 | Head of fibula-popliteal | 120 | 3.00 | 40.0 | |
| Tibial, L | ||||||||
| Ankle | 4.85 | 12.11 | 29.98 | Ankle | 4.85 | |||
| Median, R | ||||||||
| Wrist | 4.2 | 14.83 | 49.57 | Wrist | 4.20 | |||
| Elbow | 9.12 | 15.54 | 53.41 | Wrist-elbow | 240 | 4.92 | 48.8 | |
| Ulnar, R | ||||||||
| Wrist | 3.42 | 8.26 | 21.72 | Wrist | 3.42 | |||
| Below-elbow | 8.49 | 6.21 | 19.41 | Wrist–below-elbow | 215 | 5.07 | 42.4 | |
| Above-elbow | 10.53 | 5.91 | 17.33 | Below-elbow–above-elbow | 100 | 2.04 | 49.0 | |
| F-wave study | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nerve | Stim. site | F-Lat. | F-Lat. N.D. | M Lat. | F-M Lat. | F-Occurr. | Distance | FWCV | N.D. | |
| Tibial | R | Ankle | 63.8ms | 5.8ms | 58ms | 9/10.90% | ||||
| Tibial | L | Ankle | 59.9ms | 8/10.80% | ||||||
| Median | R | Wrist | 31.9ms | 3.85ms | 28.05ms | 9/10.90% | ||||
| Ulnar | R | Wrist | 32ms | 10/10.100% | ||||||
| Sensory nerve conduction study | ||||||||
|---|---|---|---|---|---|---|---|---|
| Site | Latency (ms) | Amplitude (uV) | Area (uVms) | Segment | Distance (mm) | Interval (ms) | NCV (m/s) | NCV N.D. |
| Sural, L | ||||||||
| Sural | 3.1 | 6.14 | 0.33 | Sural | 130 | 3.10 | 41.9 | |
| Sural, R | ||||||||
| Sural | 3.18 | 6.44 | 0.49 | Sural | 130 | 3.18 | 40.9 | |
| Sural | 3.18 | 5.82 | 0.26 | Sural | 130 | 3.18 | 40.9 | |
| Median, R | ||||||||
| D2-wrist | 2.76 | 14.60 | 0.88 | D2-wrist | 140 | 2.76 | 50.7 | |
| Ulnar, R | ||||||||
| Wrist-D5 | 2.18 | 24.90 | 1.51 | Wrist-D5 | 115 | 2.18 | 52.8 | |
Gibbons and Freeman2 studied a cohort of 16 patients with TIDN, providing a detailed description of the symptoms associated with the disease, with special emphasis on pain characteristics, autonomic symptoms, intra-epidermal biopsy results, and long-term follow-up. The authors show that TIDN may occur after rapid glycaemic control with insulin, as in the case presented here, and in patients receiving oral antidiabetics; this explains why the researchers prefer the term TIDN over insulin neuritis. Another study by the same authors3 proposes a set of diagnostic criteria for TIDN, including: (1) acute onset of neuropathic pain or autonomic symptoms, (2) a decrease in HbA1c level of over 2% in 3 months, and (3) onset of neuropathic pain and/or autonomic symptoms within 8 weeks of the decrease in HbA1c level.
Our patient meets the diagnostic criteria proposed by Gibbons and Freeman. Firstly, the patient experienced acute onset of pain and autonomic symptoms in the form of sustained tachycardia throughout hospitalisation and diastolic hypertension; anorexia and postprandial fullness may also be considered autonomic symptoms. Orthostatic hypotension and vasovagal syncope are the most frequently described autonomic symptoms;3,4 these were not recorded in our patient, probably due to the low clinical suspicion during hospitalisation. Secondly, he showed a 5.9-point decrease in HbA1c in less than 2 months. And thirdly, symptoms appeared following glycaemic control.
TIDN should be distinguished from other acute neuropathies presenting in these patients, especially diabetic neuropathic cachexia,5 which may be associated with similar clinical manifestations: subacute, symmetrical sensory, and motor alterations in varying degrees, associated with autonomic dysfunction. Diabetic neuropathic cachexia is characterised by anorexia, involuntary, severe weight loss, and emotional instability; these symptoms resolve completely with weight gain. The pathophysiology of diabetic neuropathic cachexia is not fully understood, and no clear diagnostic criteria have been established; the literature only includes isolated cases. We cannot rule out the possibility that our patient was affected by both conditions, given the marked involuntary weight loss and emotional lability, which was initially thought to be a response to pain.
In conclusion, TIDN or insulin neuritis is a rare, little-known entity requiring a high level of suspicion. The condition may be prevented by administering a less aggressive treatment for glycaemic control. Although TIDN is self-limiting, pain may be disabling, leading to hospitalisation.
Please cite this article as: Hernández RC, Galindo AS, Acebes EM. Neuropatía inducida por el tratamiento de la diabetes o neuritis insulínica. Neurología. 2018;33:616–618.
