



Neurally-mediated syncope (NMS) is defined as a transient loss of consciousness due to an abrupt and intermittent drop in blood pressure (BP).
ObjectivesThis study describes the putative pathophysiological mechanisms giving rise to NMS, the role of baroreflex (BR), and the interaction of its main haemodynamic variables: heart rate (HR) and BP.
DevelopmentEpisodic dysregulation affects control over the haemodynamic variables (HR and BP) mediated by BR mechanisms. During active standing, individuals experience a profound transient drop in systolic BP (SBP) due to the effect of gravity on the column of blood and probably also because of reflex vasodilation. Abnormalities in the BR in NMS could be due to a more profound drop in BP upon standing, or to delayed or incomplete vasoconstriction resulting from inhibited or delayed sympathetic activity.
ConclusionsSympathetic hyperactivity is present in patients with NMS at rest and before syncope. During active standing or passive tilting, excessive tachycardia may be followed by bradycardia and profound hypotension. Recovery of SBP is delayed or incomplete.
El síncope neuralmente mediado (SNM) se define como una pérdida súbita y transitoria del estado de alerta debido a una caída brusca de la presión arterial (PA).
ObjetivosDescribir los mecanismos putativos fisiopatológicos responsables del SNM, el papel del barorreflejo (BR) y la interacción de sus variables hemodinámicas principales: frecuencia cardiaca (FC) y PA.
DesarrolloExiste una desregulación episódica en el control de las variables hemodinámicas (FC y PA) mediadas por el barorreflejo. Durante la bipedestación activa existe una caída profunda y transitoria de la PA sistólica (PAS) debida a la acción de la gravedad sobre la columna de sangre y probablemente también a una vasodilatación refleja producida por inhibición del reflejo vasosimpático. Las anormalidades del BR en el SNM pueden ser debidas a una mayor intensidad de la caída de la PA al ponerse de pie o a una vasoconstricción retardada o incompleta debido a un reflejo vasosimpático insuficiente o retardado.
ConclusionesLos pacientes con SNM tienen en reposo y antes del síncope un estado de hiperactividad simpática. Durante el ortostatismo activo o la inclinación pasiva hay taquicardia excesiva seguida de bradicardia e hipotensión severa. La recuperación de la caída de la PAS está retardada o incompleta.
Neurally-mediated syncope (NMS) is defined as a sudden and transient loss of consciousness with spontaneous recovery. It is due to generalised brain hypoperfusion resulting from an abrupt and intense drop in blood pressure (BP).1 Initial orthostatic hypotension is defined as a short episode (20-30s) occurring 5 to 10seconds after standing involving an immediate decrease in systolic BP (SBP) of ∼40mmHg, and/or diastolic BP of ∼20mmHg.2,3 It is very frequent among young patients and is reported as a cause of syncope in 3.4% of those affected. It has the highest incidence among all causes of situational fainting.4 Prevalence of syncope is high in the general population; its incidence shows a bimodal distribution with peaks during adolescence and in patients older than 25. It is slightly more prevalent in women, with a peak of 47% vs 31% in teenage boys.5,6
The Framingham study reported an incidence of syncope which increased in patients older than 70 years, both men and women.7 Incidence rose from 5.7 per 1000 person-years in 60 to 69-year-old men to 11.1 in 70 to 79-year-old men.8,9 However, in elderly patients, cumulative incidence on syncope is more difficult to obtain, since collecting data is more complicated.7,8
In vasovagal or NMS, there is an intermittent and sudden dysregulation in the activity of the autonomous nervous system which causes a drop in BP, heart rate (HR), and brain perfusion.10
Orthostatic BP decreases with age, and this occurs in 14% to 20% of all elderly patients.2,9 Its pathophysiology has shown to be multifactorial, with baroreflex (BR) dysfunction being the most frequently mentioned putative cause.11 The array of symptoms during orthostatic hypotension manifests more frequently in young adults. This could be explained because a drop in systemic BP causes a more pronounced decline in cerebral blood flow than in elderly adults.12
Beginning in adulthood, autonomic preganglionic neurons are lost at a rate of 5% to 8% per decade. This becomes symptomatic when neuronal loss reaches 50%. In elderly adults, a head-up tilt test provokes modifications which suggest a decrease in central and peripheral autonomic reactivity; changes in BP and HR are more pronounced during active standing than during a passive tilt test. BP drops abruptly, and the increase in HR is higher; BR activity can be clearly observed, although changes are less pronounced in the elderly than in young adults. Elderly adults also display a slight or null increase in HR, an early decrease in BP, and delay in the increase in peripheral resistance and HR recovery, all of which increases the likelihood of vasovagal syncope in this age group.13
Furthermore, humoral response to orthostasis changes with age. The renin-angiotensin system seems to be less active in BP regulation during orthostasis, although resting catecholamine values seem to be higher in elderly adults. Similar increases have been observed in young adults in response to orthostasis.13
NMS is clinically characterised by initial prodromal symptoms which can manifest up to one minute before the event and include diaphoresis, pallor, nausea, abdominal discomfort, and yawning; these are followed by visual or auditory symptoms and difficulty concentrating, among others.10,14
NMS is a significant cause of morbidity and is responsible for 1% to 2% of all emergency department consultations.15 Its hospital costs in the United States amount to 2.4 billion US dollars per year.16 Scores for NMS on quality of life scales indicate that the impact of the disease is similar to that caused by more severe chronic illnesses such as epilepsy.17
Diagnosis of NMS is fundamentally based on the clinical history and the physical examination, including BP measurement during orthostasis. Currently, there are 2 methods for evaluating the response to postural changes: active standing and the head-up tilt test. The sensitivity and specificity of the head-up tilt test are difficult to determine given the methodological differences in the way that it is administered. The lack of a gold standard makes it difficult to distinguish between normal and abnormal results. However, studies with healthy volunteers and patients with a typical history of neurally mediated syncope have reported a specificity of about 90% and a sensitivity ranging from 32% to 82%.18 When a facilitating agent is used, sensitivity increases while specificity decreases. Reproducibility of studies varies between 35% and 85%. In a recently published study assessing reproducibility of the head-up tilt test and using sublingual nitroglycerin as facilitating agent, the reproducibility of a test with initially negative results was 83%, whereas reproducibility of a test with initially positive results was 79%. Mean reproducibility was 77%.19
Ross et al.20 reported a syncope induced within 11minutes after active standing, with a sensitivity of 44%. Balaji et al.21 reported a sensitivity of 61%, with a test duration of 20minutes. The positive predictive value does not seem to differ between active standing and head-up tilt test (43% and 46%, respectively). Matsushima et al.22 reported a syncope induction rate of 27% with active standing and 18% using a tilt table.
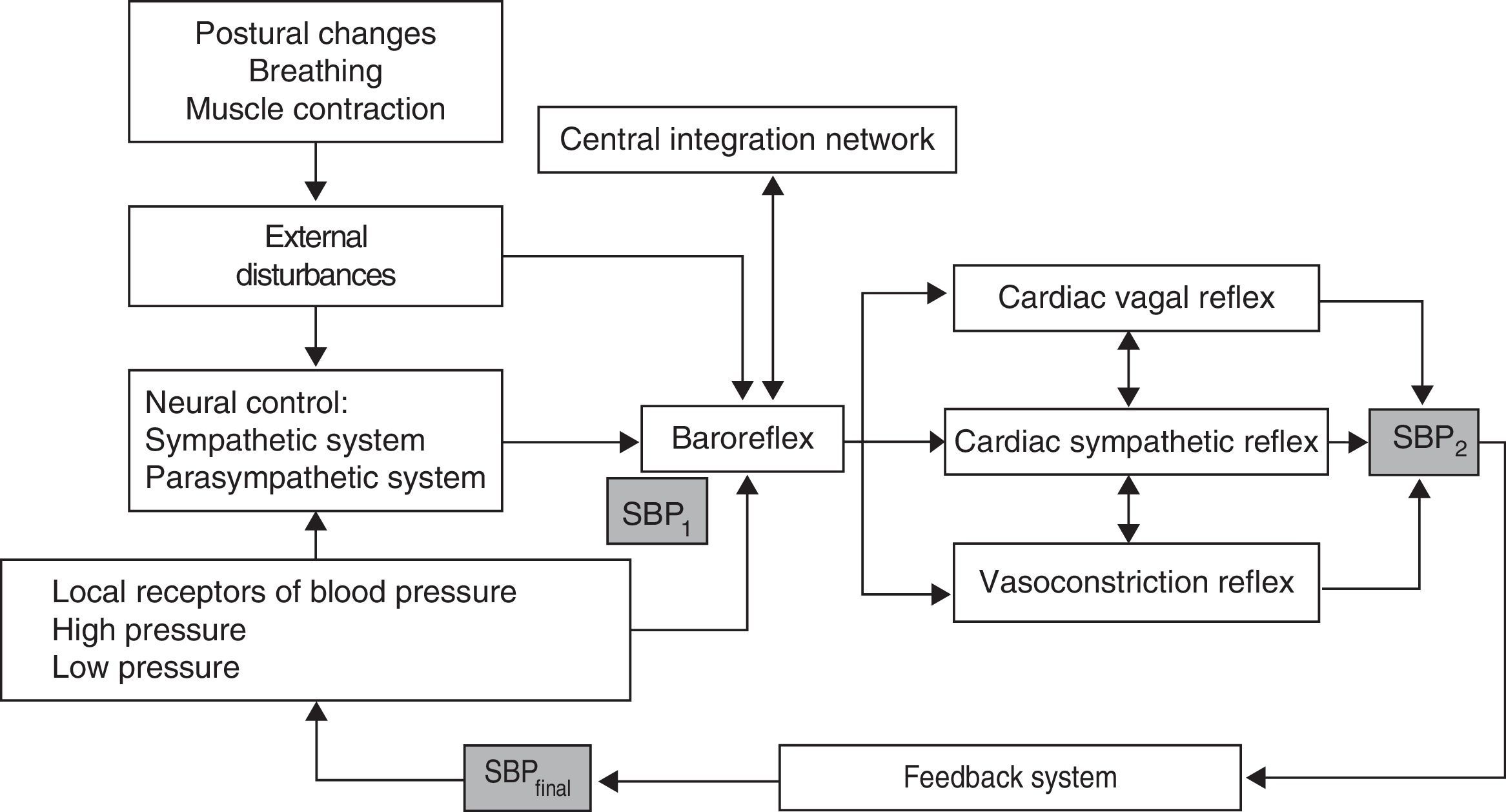
Cardiovascular system control mechanismsControl of haemodynamic fluctuations includes a number of different variables, including the following: (1) control of HR and peripheral resistance by the BR; (2) properties of the systemic arterial tree; (3) contractile properties of the myocardium, and (4) mechanical effects of respiration on BP.23
BP and HR controlControl over BP and blood flow essentially depends on 2 mechanisms (Fig. 1)24:
- 1.
Regulation of central commands: this leads to cardiovascular changes which are part of an adaptive physiological response of supranuclear mechanisms evoked by such stimuli as emotions, cognitive activity, and muscle contraction, and mediated by brain structures including the hypothalamus, the limbic system, the cingulate, and the insula; these structures act on such cardiovascular centres of the medulla oblongata as the nucleus of the solitary tract (NST) and the vagus nerve.
- 2.
BR mechanism: this mechanism enables beat-by-beat control of HR and BP in response to the stimulation of low- and high-pressure arterial receptors.

The BR controls the 2 variables determining BP: cardiac output and total peripheral resistance. In response to a drop in BP, the decrease in baroreceptor activity results in cardiac sympathetic excitation, inhibition of the cardiac vagal efferent which causes an increase in total peripheral resistance, and tachycardia with increased cardiac output. In contrast, increased BP raises baroreceptor activity, which leads to an increase in cardiac vagal activity and inhibits cardiac sympathetic and vasoconstriction activity, thereby resulting in bradycardia and hypotension.23,24
The baroreceptor reflex exerts control by activating sympathetic vasoconstrictor neurons, which are responsible for maintaining total peripheral resistance. The sympathetic efferent component of the BR is mediated by preganglionic sympathetic neurons which release acetylcholine, creating a rapid excitation of the noradrenergic postganglionic neurons innervating the vasculature responsible for peripheral resistance.24
Physiology of the BRCentral control and feedback systemThe autonomous nervous system, through its 2 branches (sympathetic/noradrenergic and parasympathetic/cholinergic), controls beat-to-beat fluctuations of the SBP. Limbic, cortical, and mesencephalic structures are responsible for immediate changes in the sympathetic tone. Adaptive physiological changes are mediated by the circulatory system, since they are part of an overall autonomic response and their response patterns depend on the central nervous system (CNS).
Barosensitive sympathetic efferents have a sympathetic tone, which is constant and is highly synchronised with changes in heart and breathing rates. These neurons are responsible for short-term regulation of SBP, as well as controlling vascular and renal systems. Furthermore, they are subject to numerous reflex regulations which respond to feedback systems: ventilation, muscle stretching, nociceptive receptors, and chemoreceptors, among others.
The rostral ventrolateral medulla has barosensitive neurons that contribute to the sympathetic tone through their response to chronotropic and inotropic impulses; they also respond to the release of neurotransmitters. The NST is the main regulatory centre of the circulatory system. It receives cardiopulmonary afferents (arterial baroreceptors, volume receptors, and peripheral chemoreceptors) and polysynaptic afferents of somatic and sympathetic afferents. Arterial baroreceptors constitute an afferent arm of the BR and are responsible for short-term regulation of the SBP.25,26
The BR is a continuous feedback loop which includes mechanoreceptors activated by the distension of the arterial wall. The increased SBP excites these baroreceptors, leading to a reflex inhibition of cardiac, renal and vasomotor efferents, which simultaneously restore baseline SBP and increase cardiac vagal activity. However, BR is able to self-calibrate to raise SBP in response to physiological changes without reducing sensitivity. This readjustment involves neural and humoral mechanisms.25–27
Feedback systemThe chronotropic effect model of the negative feedback of the BR is divided into 2 parts. The first refers to neural activity (firing frequency) in nerve terminals leading to the heart and corresponding to BP function. This effect is secondary to cardiac regulation of pressure by means of an increased stroke volume. The second part refers to the variation in HR as a function of the sympathetic and parasympathetic system.
Measurements in healthy controls show that there is a delay of some 10seconds for the maximum response mediated by the sympathetic nervous system, and a delay of less than 1second for the parasympathetic system. This may be explained by the rapid hydrolysis of the acetylcholine released by the parasympathetic system compared to the reuptake and slow depletion of norepinephrine released by sympathetic cardiac stimulation.14
The inotropic effect of BR self-regulation is less well known, but the sympathetic nervous system seems to increase systolic volume slightly alongside mean arterial pressure, within a wide physiological range. We therefore find significant differences in the delay in responses mediated by sympathetic or parasympathetic efferents. For example, given a sudden increase in BP, the parasympathetic response produces an immediate reaction (0.2-1.0s). This is the opposite of what happens in sympathetic activation at the cardiac and vasomotor level, which occurs with a delay of 2 to 3seconds, and reaches its maximum effects more slowly.26,27 Furthermore, an even slower response has been observed in reflex control of venous return.20
Other CNS structures, in addition to humoral, behavioural, and environmental factors, participate in the regulation of the cardiovascular system and contribute to the BR. Breathing, for example, interacts constantly with the modulation of the BR through its haemodynamic effects and in HR.21
BP control mechanisms are aimed at keeping BP stable, oscillating around a set point determined by physiological parameters to which the BP value converge. Fluctuations in BP and the interbeat interval (IBI) can change in the same direction (mediated by the BR) or in the opposite direction (directly mediated by the sympathetic nervous system).
Physiological effects of standing on BP and HRHuman bipedalism introduced changes and challenges in BP regulation, which evolved to meet the needs of an animal whose head was higher than its heart. Under normal circumstances, 25% of the circulating blood volume is in the thorax. Immediately after a person stands up, gravity displaces 500mL of blood to the abdomen and to the lower limbs. Approximately 50% of this volume is redistributed in a question of seconds. This process results in decreased venous return to the heart and lower cardiac filling pressures with a subsequent 40% reduction in left cardiac output. Orthostatic stabilisation is normally achieved in less than one minute. Before stabilisation, there is a decrease in BP and venous return to the right chambers, followed by the left chambers. This situation generates a sudden decrease in the unloading of high-pressure baroreceptors located in the carotid sinus and aortic arch, and low-pressure baroreceptors located in the heart and lungs. Deactivation of the baroreceptor induces tachycardia and reflex vasoconstriction to compensate the drop in BP (Fig. 2).28
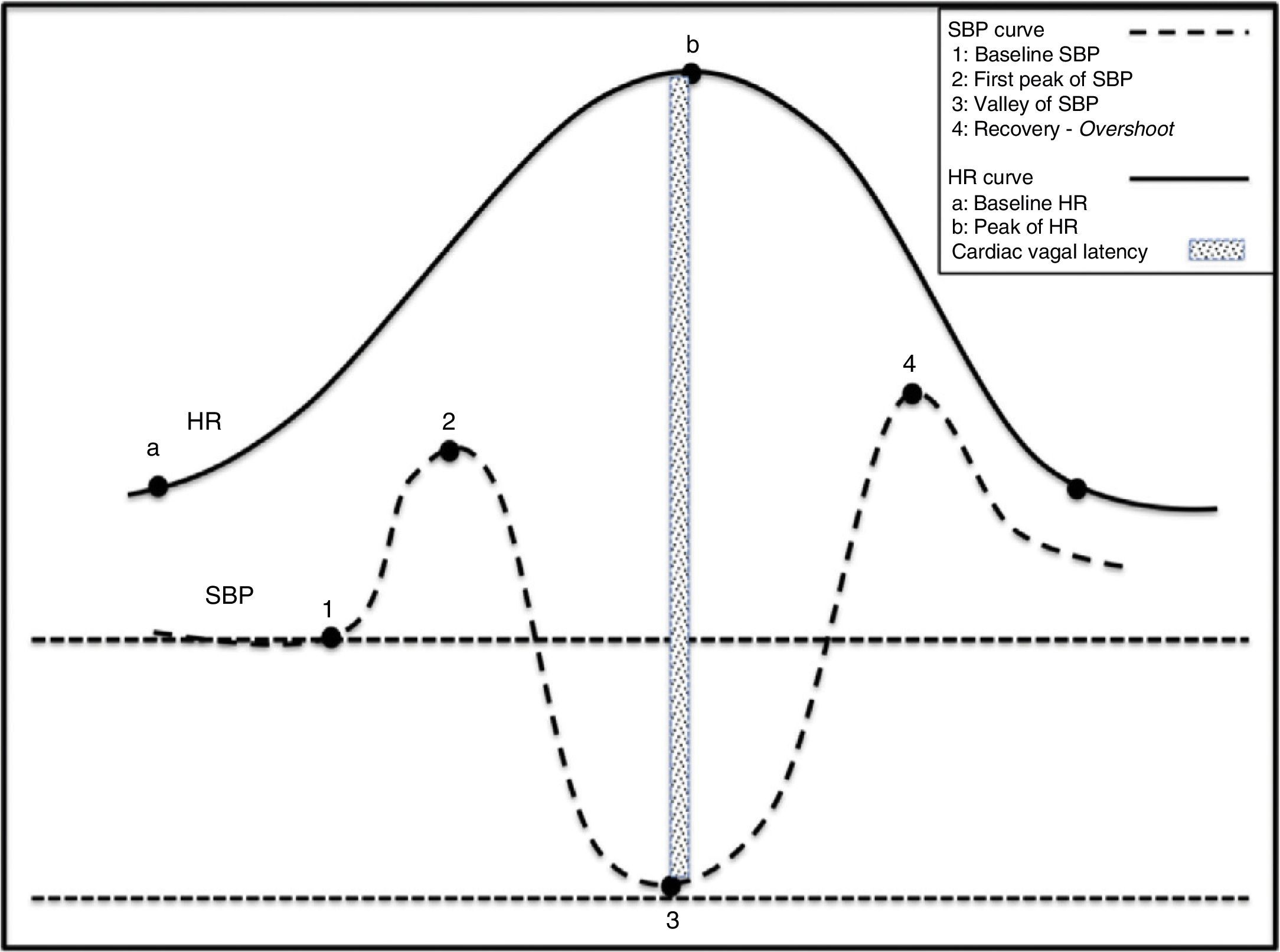
 Act of sitting generating simultaneous increases in SBP and HR. (2) Abrupt drop in SBP and rise in HR. Tachycardia is a reflex resulting from the drop in SBP. (3) Gradual increase in SBP and normalisation of HR. (4) Overshoot of SBP.")
Physiological changes in the haemodynamic variables during active standing. (1) Act of sitting generating simultaneous increases in SBP and HR. (2) Abrupt drop in SBP and rise in HR. Tachycardia is a reflex resulting from the drop in SBP. (3) Gradual increase in SBP and normalisation of HR. (4) Overshoot of SBP.
The decrease in venous outflow of approximately 40% also causes less stretching of cardiac mechanoreceptors, which are related to unmyelinated vagal afferents located in the atria and ventricles.25,28
As a result, discharge rates decrease and the change in the stimulus input to the brainstem increases sympathetic outflow, thus delivering systemic vasoconstriction. Simultaneously, decreased BP during active standing activates BP receptors in the carotid sinus, stimulating an increase in HR. This drop in SBP increases HR by 10 to 15 beats per minute and a diastolic pressure by 10mmHg, without significant changes in SBP.
Furthermore, a neurohumoral response is activated which depends on volume: the lower the volume, the higher the activation of the renin-angiotensin system.
Dysfunction of any of these processes may result in failure to elicit normal responses to postural changes. The subsequent hypotension may result in brain hypoperfusion, hypoxia, and loss of consciousness.
When transitioning from a supine position to sitting, before standing, abdominal muscles and lower limbs contract, which contributes to increases in venous return and blood flow. This delivers a sudden increase in HR and BP that peaks at 12seconds (approximately 15 beats) after standing. The initial increase in HR is attributed to sudden inhibition of the vagal tone, while the subsequent more gradual increase is caused by additional vagal inhibition and increased cardiosympathetic activity. The initial tachycardia is explained by an exercise reflex29 probably evoked by an integration of afferent signals derived from the muscle contraction with signals from CNS structures such as the insula and cingulate cortex. The resulting response is directly proportional to the intensity of the exercise. Deactivation of BR by transient hypotension causes the subsequent increase in HR.30 Immediately after the subject is seated, the BR is deactivated because if not, hypertension would abolish reflex tachycardia. That this does not occur is probably the result of central command. Following this initial increase in pressure mediated by tachycardia, healthy individuals experience a sudden drop in BP. This decrease is believed to be due not only to gravity but also to a reflex mechanism of vasodilation caused by the deactivation of low-pressure cardiopulmonary baroreceptors. HR and BP then return to a new baseline in approximately 30seconds.31 This process is shown in Fig. 2.
This physiological adaptation to standing shows that BR is able to adapt quickly to postural stress under normal circumstances.32,33
In short, several physiological mechanisms have been suggested to explain this phenomenon, and they are related to the pathophysiology of syncope during active standing. These mechanisms are:
- 1.
Muscle contraction with activation of muscle reflexes.
- 2.
Presence of local vasodilation mediators.
- 3.
Deactivation of the sympathetic system mediated by cardiopulmonary receptors with vasoconstriction inhibition and deactivation of baroreceptors: first the low-pressure cardiopulmonary receptors and then the high-pressure carotid and aortic receptors.
- 4.
Effect of gravity when standing: decreased venous return and low cardiac output.
During active standing there is a drop in pressure in the right atrium, in venous return, and in cardiac output due to the effect of gravitational acceleration on the arterial and venous blood column. When the blood in the lower limbs is driven downwards, the lower half of the body is exposed to a negative pressure of −10 to −15mmHg. This drop in central venous pressure is not immediately associated with tachycardia, but BP remains associated with vasoconstriction which can be measured in the forearm. This suggests that other reflexes contribute to circulatory homeostasis, such as cardiopulmonary low-pressure receptors, in addition to the response of the different vascular beds to this reflex.34,35
In addition, the increase in atrial pressure due to the rise in venous outflow under certain circumstances, such as lying down, raising the legs, or when inhaling suddenly, is also known to increase BP, sometimes by up to 75%. Part of this increase (15%) is due to the effect of the increase in volume on the sinus node. The remaining fraction is caused by the Bainbridge reflex. In this reflex, stretch receptors located in the atria transmit afferent signals to the brainstem through the vagus nerves. Efferent signals are then transmitted through the vagus and sympathetic nerves to generate tachycardia and inotropism.
Behaviour of BR during vasovagal syncopeIn NMS sympathetic tone increases for a time before suddenly decreasing, which causes hypotension and paradoxical vasodilatation. Cardiac vagal activation mediated by cardiac sympathetic deactivation causes bradycardia. At the same time, cardiac sympathetic activity ceases accompanied by drops in peripheral resistance and therefore in SBP. If brain perfusion drops sufficiently, loss of consciousness will occur.
The mechanism responsible for vasodilatation and bradycardia has long been assumed to be excessive stimulation of mechanoreceptors mediated by intense contraction of an insufficiently full left ventricle, which transmits paradoxical signals to the CNS (Bezoldt-Jarisch reflex). However, current evidence points to other mechanisms, such as aberrant autonomic regulation, presence of endogenous vasodilators, functional impairment of the BR, and paradoxical regulation of the CNS. All of the above may play a fundamental role and thus become the targets of future therapeutic approaches.
It has recently been demonstrated that patients with NMS show an increase in both central and peripheral vascular sensitivity to CO2, which could partially explain why some individuals are more susceptible to this phenomenon than others.36
Although we have yet to pinpoint the factors contributing to the difference between normal baroreceptor response to standing and the response during syncope induced by head-up tilt test or active standing, haemodynamic stress may play a fundamental role.
Neurohumoral factors also determine BR sensitivity in vasovagal syncope. A large neurohumoral release occurs during syncope, despite the concomitant lack of sympathetic influx.37 Four stages describe the behaviour of haemodynamic variables and, consequently, of the BR in moments immediately prior to developing NMD: the initial compensation stage refers to transient hypotension; it returns to baseline values in approximately 30seconds, with diastolic pressure remaining above baseline. In the second stage, tachycardia arises concomitantly and subsequently persists for a mean duration of 4minutes; this responds to sudden inhibition of cardiovagal activity. This blockade is attributed to the exercise reflex. Patients developing NMS exhibit a third stage of instability with oscillating HR and BP. Although HR increases, this rise does not reach previous values, just as the drop in BP does not reach the values measured in the compensation stage. This stage is related to the onset of the clinical symptoms of presyncope, and its approximate mean duration is 2.1minutes. Finally, the patient develops the presyncope which is haemodynamically associated with a sudden drop in BP and bradychardia, with the subsequent recovery or development of syncope.37
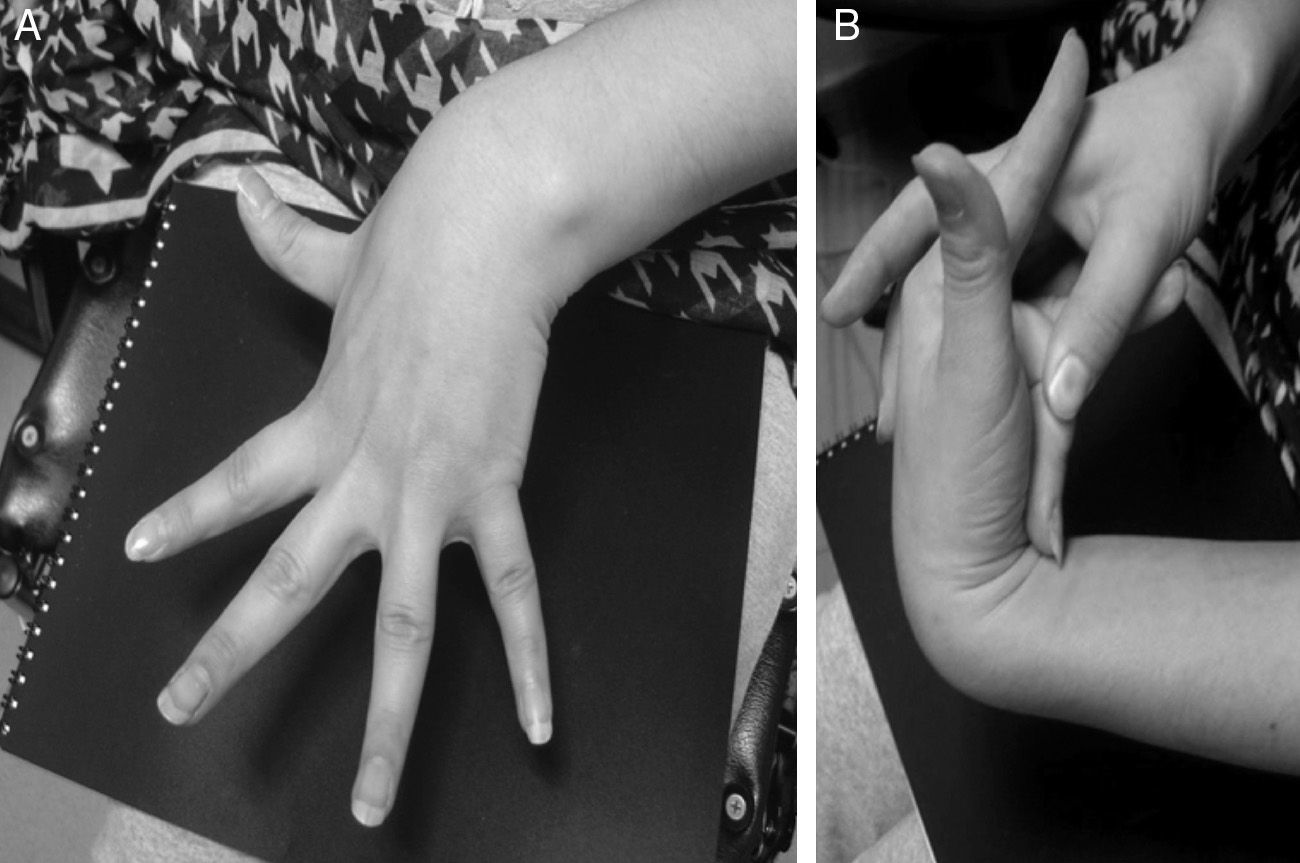
An additional mechanism, which could explain the role of dysautonomia in the genesis of NMS and was initially described in patients with postural orthostatic tachycardia syndrome (POTS), is the regional autonomic denervation usually found in the lower limbs (neuropathic theory). This denervation could be responsible for abnormal responses to the Valsalva manoeuver, such as a less marked increase in BP due to a decreased vasoconstriction sympathetic response. A decrease in the secretion of acetylcholine in response to physiological manoeuvers and pharmacological stimuli has also been described.38 In addition, some patients present pooling of the blood in the lower limbs while standing, which may be explained by excessive compliance and increased capacity of the venous vasculature during orthostasis. This situation might explain why syncope incidence increases in patients with hypermobility syndromes or Ehlers–Danlos syndrome type III (Fig. 3).38
 Hyperextension of fingers and wrists. (B) The hypermobile middle finger can be pressed to the forearm.")
BR has at least 2 afferents: high-pressure receptors located on the carotid and aortic arteries, and the low-pressure receptors, located on the right chambers of the heart and the pulmonary circulation. It also has 3 efferents: cardiac vagal reflex, cardiac sympathetic reflex, and vasoconstriction reflex.26,30,38–41 In addition, there is a central integration network located on the NST, the nucleus ambiguus, the ventrolateral region of the medulla, and the intermediolateral column of the spinal cord.40,42 The BR is a polysynaptic reflex affected by such supranuclear structures as the hypothalamus, the limbic system, and the insular cortex.26,39,43 This supranuclear influence is not well understood yet, but it is associated with alterations in BP attributed to mechanisms or central commands, and linked to emotions, cognitive activities, and voluntary muscle contraction.26,38,43 The aim of the BR is short-term stabilisation of BP in response to the different variations generated by the internal or external stimuli to which it is constantly exposed.39 The 2 main external changes inducing haemodynamic alterations are gravitational stress and respiratory movements (Fig. 1).42,44,45
Active standing generates an abrupt drop in BP (40mmHg) during the first 30seconds, in addition to a significant increase in HR (∼15 beats) and cardiac output accompanied by a significant decrease in peripheral resistance.22 A pronounced decrease in BR sensitivity has been demonstrated in patients with NMS. The analysis of spontaneous HR fluctuation suggests that patients with poor orthostatic tolerance present prolonged latency of the cardiac response to BR. Analysis of healthy subjects shows that BR sensitivity decreases proportionally to the angle of inclination of the body, and that dynamic adaptation of BR to a physiological alteration occurs rapidly. BR latency is proportional to the angle of inclination, and this indicates that functional impairment of the BR results from reduced vagal activity and increased sympathetic tone.32,46 Therefore, vagal changes mediated by BR and induced by postural changes, in addition to atropine's vagotonic and vagolytic effects, are known to be associated with a delay between baroreceptor stimulus and response to HR.46,47
In patients with vasovagal syncope, the weakened cardiac sympathetic and vasosympathetic reflexes, with the resulting haemodynamic instability, show a significant and independent association with syncope recurrence.47
ConclusionsNeurally mediated syncope is a frequent complication that can present at any age. It is both genetic and familial and has been observed in patients with Ehlers–Danlos syndrome type III with joint hypermotility. It generally presents in individuals with no cardiac or neurological disease. From a pathophysiological viewpoint, NMS is characterised by an initial state of sympathetic hyperactivity which translates clinically into tachycardia, sweating, tremor, and cutaneous vasoconstriction. At the time syncope occurs, sympathetic hyperactivity, both at the heart and resistance vessel levels, stops abruptly. There is an increase in cardiac vagal activity which can cause asystole or marked bradycardia; furthermore, deactivation of sympathetic activity to the blood vessels (vasoconstriction reflex) can elicit pronounced vasodilation with severe hypotension. The intermittent nature of NMS continues to pose challenges and makes it difficult to study. The most plausible hypothesis hinges on the presence of an intermittent dysfunction of BR control over BP and HR, which can occur at the peripheral or the central level.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Malamud-Kessler C, Bruno E, Chiquete E, Sentíes-Madrid H, Campos-Sánchez M. Fisiopatología del síncope neuralmente mediado. Neurología. 2016;31:620–627.
