



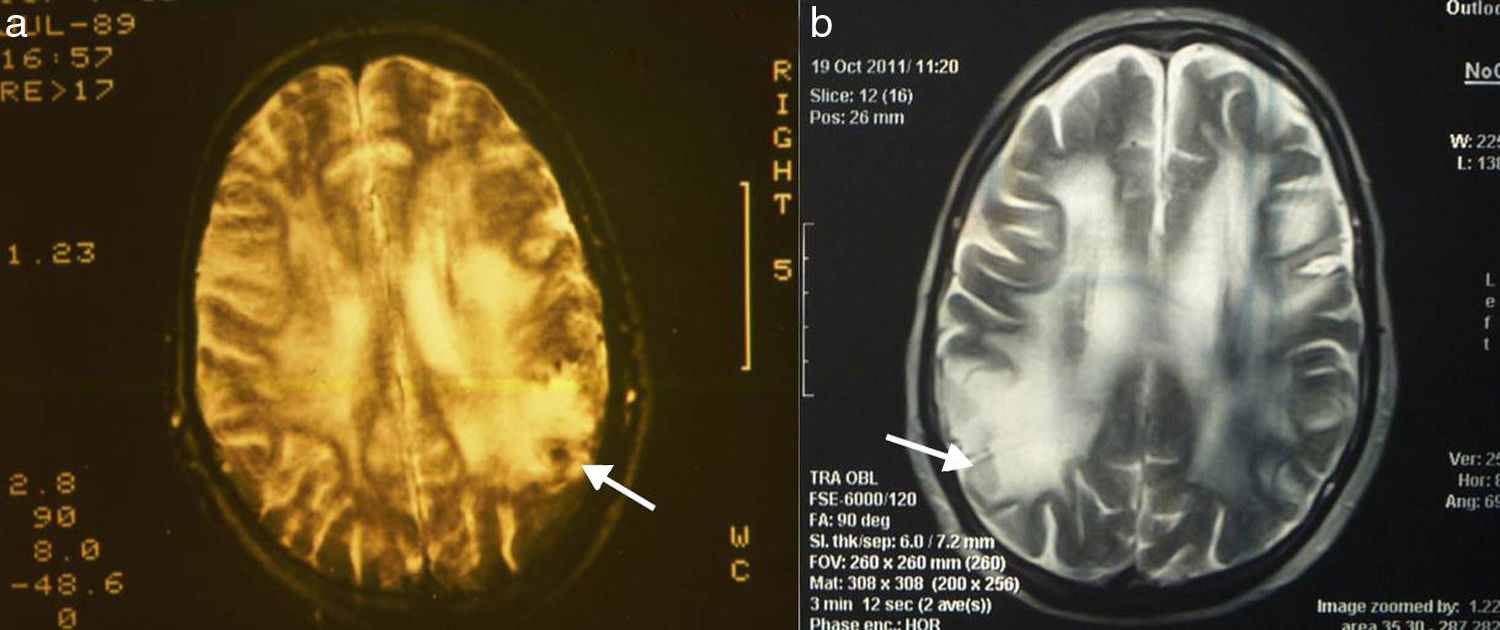
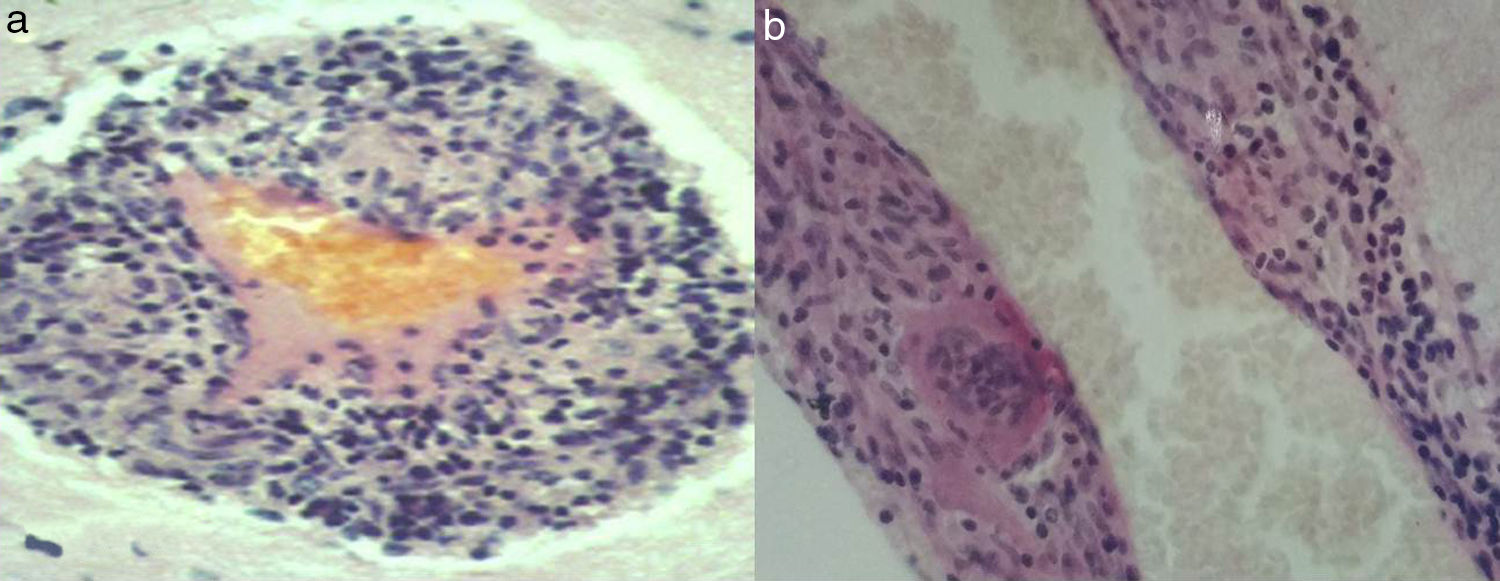
The central nervous system (CNS) may develop many forms of angiitis; some are multisystemic whereas others are primary. Primary angiitis of the central nervous system (PACNS), first described in 1959,1 is a form of vasculitis restricted to the small arteries of the meninges and parenchyma. This rare disease causes immune-mediated inflammation. Typical manifestations include headache, delirium, small-vessel stroke, and progressive dementia. Brain magnetic resonance imaging (MRI) shows multiple ischaemic foci in both cerebral hemispheres. Cerebrospinal fluid (CSF) analysis shows abnormal protein levels and cell counts. Cerebral angiography shows signs of angiitis in 50% of cases.2 Diagnosis is based on clinical, MRI, CSF, angiography, and brain biopsy findings, after ruling out systemic inflammation or infection. Histological findings include lymphocytic, granulomatous, histiocytic, or eosinophilic infiltrates, or signs of necrosis.3,4 We describe the extended follow-up of a patient with PACNS, with clinically satisfactory outcomes. MR images revealed extensive leukoaraiosis, which remained stable over the course of 25 years. The patient was continuously treated with methylprednisolone and received cyclophosphamide periodically. In March 1989 our patient, a 42-year-old woman with no relevant medical history, began to experience persistent headache, nausea, and vomiting. A week later, she also displayed mental confusion and gait instability, which led to her admission to hospital. During the examination, the patient was disoriented with regard to time, space, and person; she was drowsy and showed moderate left-sided hemiparesis and neck stiffness. A head computed tomography (CT) scan revealed hypodensities in both cerebral hemispheres. A brain MRI scan confirmed extensive leukoaraiosis, predominantly affecting the right hemisphere (Fig. 1a). A blood analysis showed normal results for blood counts; erythrocyte sedimentation rate (10mm/h); glucose; kidney, liver, and thyroid function; HIV; CRP; rheumatic factors; ACE (sarcoidosis); and proteins. A CSF analysis disclosed 20cells/mm3 and 160mg proteins; glucose levels were within normal ranges, and the patient tested negative for common pathogens, Koch bacillus, and mycosis. The PPD skin test yielded negative results. A chest, abdomen, and pelvis CT scan revealed no abnormalities. A Doppler ultrasound of the carotid arteries revealed no occlusion, and a cardiovascular examination yielded normal results. The patient had no skin lesions, mouth or genital ulcers, or uveitis; Behçet disease was therefore ruled out. Cerebral angiography revealed a narrowing of branches of the anterior cerebral artery. Given suspicion of arteritis, we performed a meningeal and brain biopsy of the right parietal cortex. The biopsy revealed inflammatory infiltrates in small vessels; cells were predominantly lymphocytes, with some histiocytes and multinucleated giant cells (Fig. 2a and b). It was not possible to typify lymphocytes in 1989. The patient started treatment with oral methylprednisolone 60mg/day, achieving an extraordinary improvement in one week. CSF was normal 2 weeks after treatment was started. At month 3, methylprednisolone dose was gradually reduced to 4-8mg on alternating days. The patient attended annual follow-up consultations, during which she was evaluated for cognitive function (MMSE, ADAS-Cog, ADCS-ADL, and CDR), mobility (Timed Up and Go test), and sphincter control. She also underwent a brain MRI scan every 2 years. Blood tests for varicella-zoster virus, Epstein–Barr virus, and cytomegalovirus performed at other centres yielded negative results. The patient was asymptomatic for 8 years; her case was presented at the World Congress of Neurology in 1997.5 In the eighth year of follow-up, the patient displayed gait instability; methylprednisolone dose was increased to 40mg/day, and symptoms disappeared within 2 weeks. Treatment with methylprednisolone was subsequently maintained at 4-8mg on alternating days; cyclophosphamide dosed at 50mg/day was added periodically for 6-month periods. The patient developed cataracts in both eyes, which were treated surgically. At 15 years of follow-up, she displayed cautious gait and had some minor falls. The patient continues to display this type of gait at present, and uses a cane outside of the home. Symptoms did not improve after increasing the dose of methylprednisolone for a month. Over 25 years of follow-up, the patient's cognitive function has remained within normal ranges; mean scores are as follows: MMSE 27.9±2.74; ADAS-Cog 11.4±3.26; ADCS-ADL 25.8±2.29; and CDR 0.41±0.04. In the last 10 years, the patient has displayed cautious gait, taking slow, short steps, with the feet barely lifting from the floor. Muscle strength, muscle tone, sensitivity, and deep tendon reflexes are normal. Abnormal gait was attributed to involvement of the frontal subcortical white matter. The mean Timed Up and Go test score was 11.5±1.01 for the first 15 years of follow-up and 19.7±1.12 for the last 10 years. Sphincter function has remained normal at all times throughout follow-up. Brain MR images revealed no changes from symptom onset to present (Fig. 1a and b). The case presented here demonstrates the variability of PACNS in terms of form of presentation, and the multiple differential diagnoses hindering aetiological diagnosis of the condition before brain biopsy can be performed. The most frequent symptoms include refractory headache, encephalopathy, and focal symptoms; spinal cord involvement is much less frequent.6 CSF study results are abnormal in nearly 80% of patients,7 with oligoclonal bands being observed in half of cases.8 Inflammatory infiltrates may contain a combination of lymphocytes, histiocytes, and multinucleated giant cells; treatment is always based around long-term immunosuppression. PACNS is a rare disease, which means that few controlled studies of specific drugs have been conducted. One of the few published controlled studies on this topic included children with PACNS who were treated with methylprednisolone infusions, followed by maintenance therapy with cyclophosphamide plus either azathioprine or oral mycophenolate mofetil. Immunosuppresion in children may improve long-term outcomes; some of the patients included in the study were successfully managed for 7 years.9 To our knowledge, ours is a unique case of long-term follow-up. Maintaining immunosuppression contributes to preserving functional independence for daily living activities. We should also highlight the abnormal persistence of leukoaraiosis on brain MR images over the course of 25 years. Although leukoaraiosis has a different pathogenic mechanism, once it appears, it persists or worsens over time.10
 (1989) T2-weighted sequence showing periventricular hyperintensities (leukoaraiosis) in the centrum semiovale; these had no mass effect and were more marked in the right hemisphere. (b) (2011) T2-weighted sequence showing similar findings. Arrows indicate the biopsy site.")
Brain MRI. (a) (1989) T2-weighted sequence showing periventricular hyperintensities (leukoaraiosis) in the centrum semiovale; these had no mass effect and were more marked in the right hemisphere. (b) (2011) T2-weighted sequence showing similar findings. Arrows indicate the biopsy site.
 and sagittal planes (b) showing small meningeal and brain arteries with inflammatory infiltration into the walls: lymphocytes, histiocytes, and multinucleated giant cells (H&E×400).")
Please cite this article as: Domínguez RO, Perdomo Rojas LM, Gonzalez SE, Bartolomé EL. Angeítis primaria del sistema nervioso central: 25 años de seguimiento y leves secuelas motoras. Neurología. 2019;34:276–278.
