



In humans and animal models, Alzheimer disease (AD) is characterised by accumulation of amyloid-β peptide (Aβ) and hyperphosphorylated tau protein, neuronal degeneration, and astrocytic gliosis, especially in vulnerable brain regions (hippocampus and cortex). These alterations are associated with cognitive impairment (loss of memory) and non-cognitive impairment (motor impairment). The purpose of this study was to identify cell changes (neurons and glial cells) and aggregation of Aβ and hyperphosphorylated tau protein in the primary motor cortex (M1) in 3xTg-AD mouse models at an intermediate stage of AD.
MethodsWe used female 3xTg-AD mice aged 11 months and compared them to non-transgenic mice of the same age. In both groups, we assessed motor performance (open field test) and neuronal damage in M1 using specific markers: BAM10 (extracellular Aβ aggregates), tau 499 (hyperphosphorylated tau protein), GFAP (astrocytes), and Klüver-Barrera staining (neurons).
ResultsFemale 3xTg-AD mice in intermediate stages of the disease displayed motor and cellular alterations associated with Aβ and hyperphosphorylated tau protein deposition in M1.
ConclusionsPatients with AD display signs and symptoms of functional impairment from early stages. According to our results, M1 cell damage in intermediate-stage AD affects motor function, which is linked to progression of the disease.
En el cerebro del humano y en el de modelos animales, la enfermedad de Alzheimer (EA) se caracteriza por la acumulación del péptido β-amiloide (βA), de la proteína tau hiperfosforilada, degeneración neuronal y gliosis astrocítica que son prominentes en regiones cerebrales vulnerables (hipocampo y corteza). Estas alteraciones se relacionan con el deterioro cognitivo (pérdida de la memoria) y no cognitivo en la función motora. El objetivo de este trabajo fue identificar en el modelo (3xTg-AD) los cambios celulares (neuronas y astroglía) y la agregación de βA y tau hiperfosforilada en la corteza motora primaria (M1) en una etapa intermedia de la EA y su relación con el desempeño motor.
MétodosSe utilizaron hembras 3xTg-AD de 11 meses de edad, comparadas con no transgénicas (No-Tg) de la misma edad. En ambos grupos, se evaluaron el desempeño motor (campo abierto) y el daño celular con marcadores específicos: BAM10 (agregados βA extracelulares), tau 499 (hiperfosforilada), GFAP (astrocitos) y Klüver-Barrera (neuronas) en la M1.
ResultadosLas hembras 3xTg-AD en etapa intermedia de la patología mostraron alteraciones motoras y celulares asociadas al depósito de βA y tau hiperfosforilada en la M1.
ConclusionesDesde etapas tempranas de la EA se observan signos y síntomas de deterioro funcional. Sin embargo, en este estudio reportamos que en la etapa intermedia de la patología se encuentran establecidas las características de daño en la M1 asociadas al desempeño motor. Eventos que se relacionan con el avance de las características clínicas de la patología.
Dementia is a neurological syndrome characterised by a significant decline in cognitive performance (amnesia), psychological alterations, and changes in executive functions, with an impact on the patient’s ability to independently perform the activities of daily living.1 Alzheimer disease (AD) is the most common cause of dementia in elderly people. The disease is heterogeneous, with clinical signs including cognitive disorders, memory loss, and behavioural alterations,2 as well as such non-cognitive symptoms as motor dysfunction with balance disorders, slow gait speed, and motor signs including loss of muscle mass and strength, leading to a progressive loss of independence3–5; these symptoms may constitute preclinical signs of AD.6,7 While decreased hippocampal volume is predictive of dementia, onset may also be focal or asymmetrical, manifesting for example as cortical atrophy, which is associated with such non-cognitive symptoms as progressive apraxias and asymmetrical brain involvement.8 Clinical assessment distinguishes 3 stages of development: a hippocampal stage (associated with amnestic deficit), followed by language disorders and apraxias (functional alterations involving the parietal, temporal, and occipital cortical association areas), and a global stage, involving the extrapyramidal pathway.8
A concomitant cellular mechanism in AD is synaptic dysfunction, which begins in vulnerable areas (the hippocampus and neocortex) affected by β amyloid (Aβ) accumulation and hyperphosphorylation of cytoskeletal tau protein.9,10 These processes cause irreversible neurodegeneration and progressive memory loss.11 The physiological functions of Aβ include controlling cholesterol transport,12 but its accumulation in oligomers of 32 to 42 amino acids can cause synaptic alterations and neuronal damage.13 Other factors associated with the disease are oxidative stress; neuroinflammation, which gives rise to Aβ plaques; and tau protein truncation and hyperphosphorylation, causing cytoskeletal changes in cells of the hippocampus,14 a site of neuronal plasticity (the process regulating memory and learning).15 Reducing tau cytotoxicity has been shown to prevent Aβ-induced memory deficits and neuronal dysfunction in transgenic mouse studies.16
Numerous mouse models of AD are currently available,17 but the triple transgenic model (3xTg-AD), which overexpresses 3 human proteins (PS1M146V, APPSwe, and tauP3001), replicates many aspects of AD progression, with deposits of Aβ (plaques) and hyperphosphorylated tau developing according to age and specific brain area. At 3 to 4 months of age, mice display cognitive impairment associated with Aβ plaques in the CA1 region of the hippocampus; extracellular deposits are observable in the frontal lobe at 6 to 12 months.18 These characteristics enable research into various potential treatments in the intermediate phases of AD progression. During progression, mice also show microglial activation and reactive astrogliosis in amyloid plaques19; astrocytes are activated in response to the damage, and glial fibrillary acidic protein (GFAP) expression increases in their intermediate filaments; such morphological changes as hypertrophy are also observed.20,21 Astrocytes dynamically provide trophic and metabolic support to neurons, modulate information processing, and regulate neuronal activity and synaptic plasticity.22–24 Sustained homeostatic dysregulation of astrocytes25 can have a severe impact on the stability of neuronal microcircuits; in AD, this dysregulation is concomitant with extracellular deposition of Aβ and hyperphosphorylated tau protein.26 This study aims to assess alterations observed in the primary motor cortex (M1 region) in the intermediate period before plaque deposition (11 months), and their possible relationship with the non-cognitive symptoms of AD (motor deficiencies) in 3xTg-AD transgenic mice.
Material and methodsAnimalsAll animals used in this study were maintained and supervised by a qualified veterinarian, in accordance with the international standards established by the National Institutes of Health and the National Academy of Science for the handling and use of experimental animals. The study was approved by the Bioethics Committee of the Institute of Neurobiology pertaining to Universidad Nacional Autónoma de México. Animals were housed 4 to a cage with free access to water and food, in optimal conditions in a vivarium (with a 12:12 light/darkness cycle, temperature of 20°C, and relative humidity of 40%-50%). The triple transgenic mouse model was developed in the LaFerla laboratory at the University of California, Irvine17; it possesses 3 human transgenes (APPSwe and tauP301L injected into PS1M146V knock-in embryos) on a 129/C57BL6 hybrid background. Non-transgenic (non-Tg) mice were from the same hybrid background. We only used female mice of 11 months of age; these hybrids of the original reproductive units were weaned at 30 days of age. Mice in the 3xTg-AD group were homozygous for AD, whereas non-Tg mice were not. All animals were housed 3 to a cage from weaning until the beginning of the experiment.
GenotypingIn all animals, DNA was extracted from the most caudal segment of the tail (0.5cm in length); tissue was placed in a 1mL Eppendorf® tube for alkaline lysis. A polymerase chain reaction study was then performed to verify the presence of the genes encoding amyloid precursor protein and tau protein (bands observed at 500 and 350bp, respectively) and presenilin 1 (observed in 2 bands, at 180 and 350bp). All gels were visualised under UV light.
Behavioural studyThe open field test is a motor test used to determine levels of spontaneous locomotor activity and exploratory behaviour in rodent models of central nervous system disorders.17 Eight animals were included in each group; each animal was placed for 15minutes in a rectangular arena (45×45cm), with the experimenter out of sight. The floor of the arena was uniformly lit and marked to show the separation between central and peripheral areas; the animals’ movement was recorded with a digital camera mounted immediately above the arena. The ANY-Maze video tracking software (v 4.82; Stoelting Co., USA) was used to track activity; distance travelled in centimetres and time spent travelling were considered as measures of locomotor activity.
Fixing and histological processingSix animals from each group were randomly selected for histological study. Subjects were anaesthetised with intraperitoneal pentobarbital (300μg/kg body weight) and euthanised with a transcardial perfusion of 4% paraformaldehyde in buffer solution (pH 7.4, 0.1M) and 1000U of 0.1% heparin and procaine. Brains were removed and fixed in 4% paraformaldehyde for 24hours at room temperature. The hemispheres were separated and cut into 50μm sagittal slices with a vibratome (Vibratome® 3000 Deluxe tissue sectioning system); we used the coordinates 1.92 to 2.28mm lateral to bregma, as indicated in the Paxinos and Franklin27 mouse brain atlas. One hemisphere was processed using the conventional Klüver-Barrera technique for the morphometric analysis of neurons; samples were visualised with light microscopy (Nikon Eclipse Ci-S/Ci-L, equipped with a DS-U3 camera and 100× plan apochromat objective lens [Plan-Apochromat, 1.25 NA/160]); equidistant and equivalent sections from both experimental groups were studied. We studied cortical areas of 136×581μm from the M1 region, using the Q-Win and ImageJ computer programs (National Institutes of Health, USA) to quantify normal and damaged neurons. The criteria for neurons to be considered damaged included cytoplasmic hyperchromasia, cellular shrinking, and nuclear fragmentation; these counts were expressed as the number of neurons within a defined area. For the purposes of comparison at the interstitial level, all counts are expressed as the number of neurons in an area of 0.1mm2.
Immunohistochemical study of Aβ and hyperphosphorylated tau proteinA second series of sagittal slices was used in a free-floating immunohistochemical study (n=6 per group). Mouse anti-Aβ antibodies were stained using Bam10 (monoclonal IgG; Sigma-Aldrich, no. A5213, 1:1000) as the primary antibody and a biotinylated secondary antibody (anti-mouse IgG; Vector, 1:500). Human tau protein was stained using anti-human PHF-tau (monoclonal IgG; Thermo Scientific, 1:200) as the primary antibody and a biotinylated secondary antibody (anti-mouse IgG; Vector, 1:500). ABC substrate and peroxidase kits were used to visualise antibody binding. Images were recorded using a Nikon Digital Sight camera (DS-Fi2/DS-Fi1/DS-Vi1) with a 40× objective lens (Plan-Apochromat, 0.65 NA/160).
Immunohistochemical analysis of astrocytesGFAP expression was identified by fluorescence; tissues were incubated in 0.1M buffer solution with 0.1% triton for 30minutes, then incubated again in 0.1M buffer solution with 10% normal goat serum and 0.1% triton with agitation for one hour. The solution was removed and the tissue was incubated with an anti-GFAP primary antibody (rabbit polyclonal antibody; Dako, 1:500) with agitation, overnight at 4°C. After 24 hours’ incubation with the primary antibody, this was removed and the tissue was washed 5 times in 0.1M buffer solution for 8minutes each time; samples were then incubated with the secondary antibody conjugated to Alexa Fluor 488 dye (Invitrogen; 1:500) for 2hours at room temperature with constant agitation. Samples were then washed 5 times with 0.1M buffer solution for 8minutes each time and mounted on slides with Vectashield antifade mounting medium; cover slips were sealed with nail varnish.
We evaluated immunoreactivity and quantified GFAP-positive cells using a confocal microscope (Leica TCS SP2) with a 100× plan apochromat objective lens (Plan-Apochromat; 2.25 NA/160); in the M1 region, we studied an area of 136×581μm. Images were analysed with the ImageJ software and results are expressed as arbitrary units across a 0.1mm2 area.
Morphological changes in astrocytic processes were measured with a modified Sholl analysis,28 comprising 6 concentric circles at 10μm of separation. The central circle was placed over the centre of the astrocyte and we quantified intersections between processes and the concentric circles. For each experimental group, we selected all complete astrocytes in the M1 region in each hemisphere; this approach enabled us to compare cell morphology by observing astrocytic immunoreactivity.
Statistical analysisOpen field test results were analysed using the t test; the t test and ANOVA were used to analyse results from morphometric and immunohistochemical studies. Statistical analysis was performed using the StatView and GraphPad software, and results are expressed as the mean and standard error of the mean for the 6 samples in each group; the threshold for statistical significance was set at P≤.05.
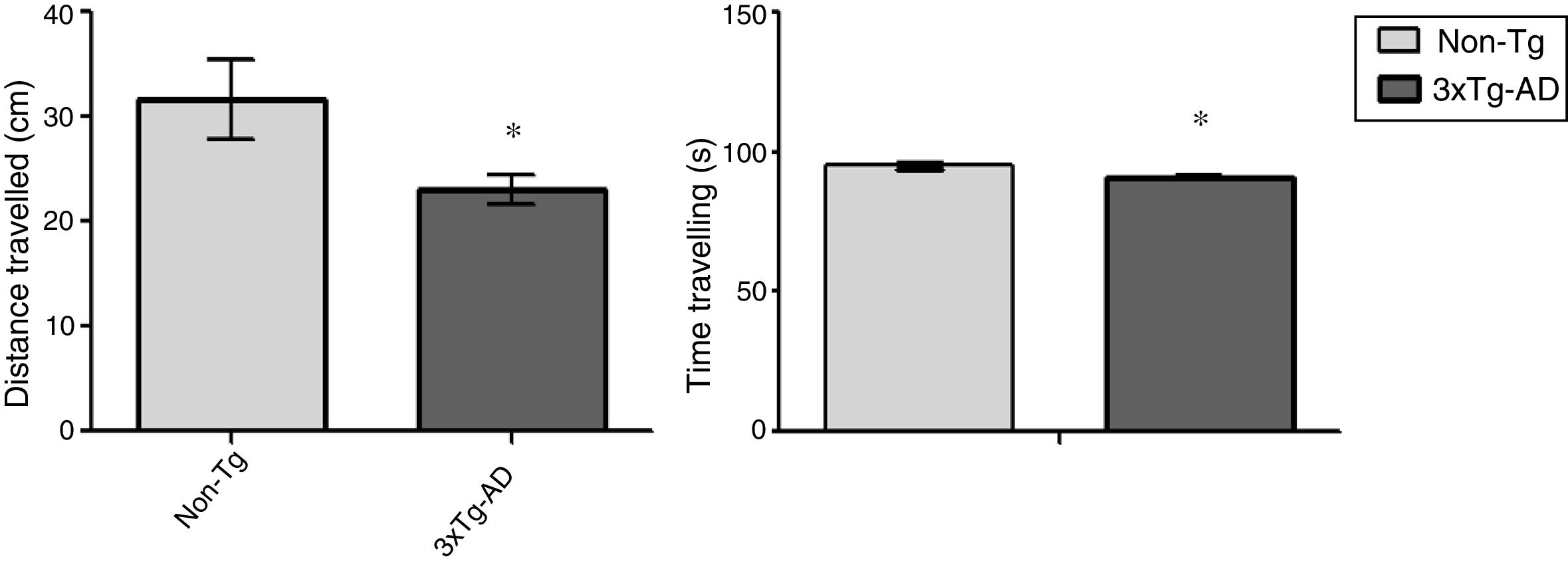
ResultsOpen-field testLocomotor activity was significantly reduced in 3xTg-AD mice compared to non-Tg mice (P<.05) in the 15-minute motor test. Motor activity was measured in centimetres travelled; 3xTg-AD mice covered a shorter distance and spent less time travelling that the non-Tg group. Time spent immobile was not considered in this analysis (Fig. 1).
.")
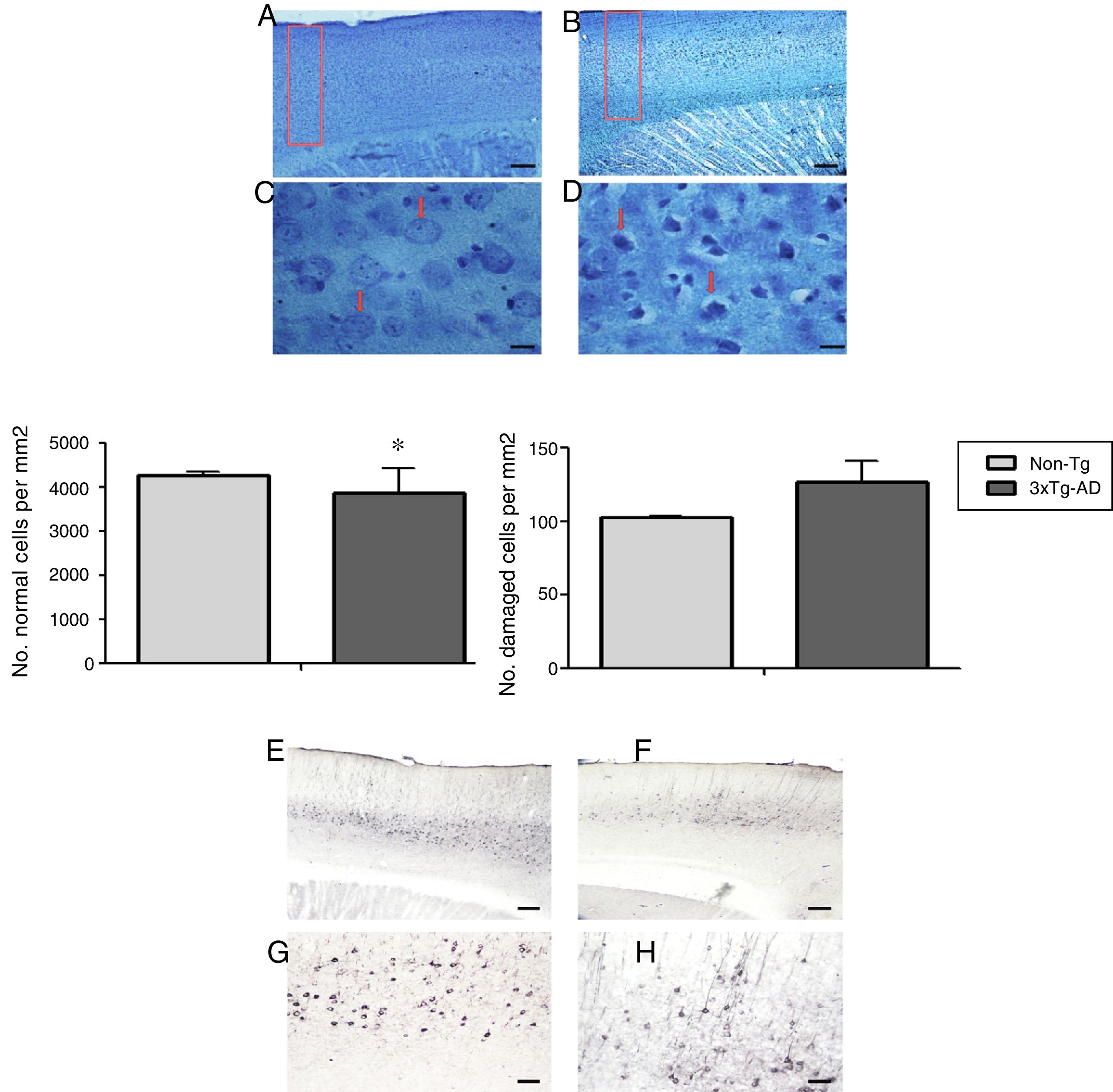
Morphometric analysis of the M1 region from 3xTg-AD mice revealed a significant reduction in the number of normal neurons and a non-significant increase in the number of abnormal neurons vs non-Tg mice (Fig. 2) in slices stained with the Klüver-Barrera technique. In the same area, qualitative observation of immunohistochemical labelling showed extracellular aggregates of Aβ and hyperphosphorylated tau (Fig. 2).
 Sagittal sections stained using the Klüver-Barrera technique; the arrows indicate cells with normal morphological characteristics (non-Tg). B and D) Representative tissue sample from the 3xTg-AD model; the arrows indicate cells with signs of damage. The graphs show mean±standard error of the mean (6 individuals per group) for the numbers of normal and abnormal cells. The asterisk indicates a statistically significant difference. E and G) Motor cortex (M1 region) from a 3xTg-AD mouse; Aβ expression is labelled with antibody Bam10. F and H) Labelling of tau protein. Calibration bars: 100μm (A, B, E, and F) and 10μm (C, D, G, and H).")
Representative photomicrographs from the primary motor cortex. A and C) Sagittal sections stained using the Klüver-Barrera technique; the arrows indicate cells with normal morphological characteristics (non-Tg). B and D) Representative tissue sample from the 3xTg-AD model; the arrows indicate cells with signs of damage. The graphs show mean±standard error of the mean (6 individuals per group) for the numbers of normal and abnormal cells. The asterisk indicates a statistically significant difference. E and G) Motor cortex (M1 region) from a 3xTg-AD mouse; Aβ expression is labelled with antibody Bam10. F and H) Labelling of tau protein. Calibration bars: 100μm (A, B, E, and F) and 10μm (C, D, G, and H).
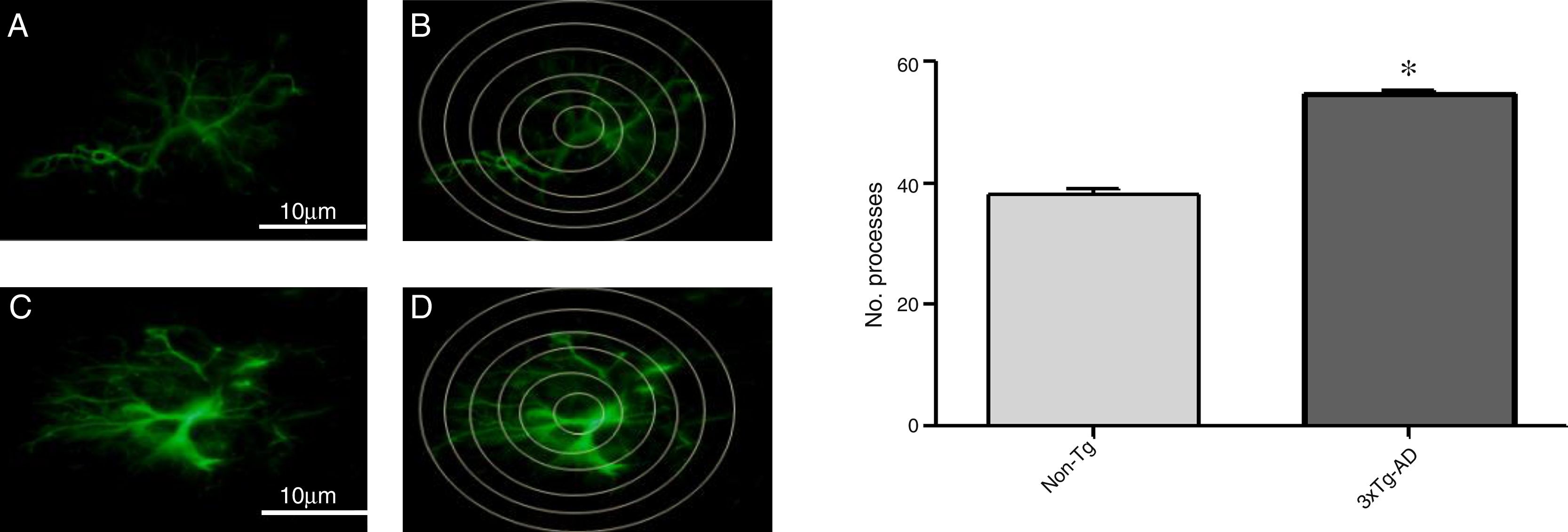
Quantitative analysis of intersections of GFAP-immunoreactive astrocytes in the primary motor cortex
Increased GFAP immunoreactivity was observed in the M1 region of 3xTg-AD mice compared to non-Tg mice. GFAP-positive astrocytes showed signs of reactivity, hypertrophy, lengthening of the cell body, and increased volume and surface area. Fig. 3 illustrates the increased number of intersections of the processes of GFAP-immunoreactive astrocytes (modified Sholl analysis) in 3xTg-AD mice compared to the non-Tg group.
 Photomicrographs showing GFAP immunoreactivity in the M1 region of 11-month-old female non-Tg and 3xTg-AD mice, respectively. B and D) Sholl analysis of GFAP-immunoreactive cells from the M1 region of non-Tg and 3xTg-AD mice, respectively. Graphs show mean±standard error of the mean (6 individuals per group) for the number of GFAP-immunoreactive processes intersected by the concentric circles for each group. The asterisk indicates a statistically significant difference.")
A and C) Photomicrographs showing GFAP immunoreactivity in the M1 region of 11-month-old female non-Tg and 3xTg-AD mice, respectively. B and D) Sholl analysis of GFAP-immunoreactive cells from the M1 region of non-Tg and 3xTg-AD mice, respectively. Graphs show mean±standard error of the mean (6 individuals per group) for the number of GFAP-immunoreactive processes intersected by the concentric circles for each group. The asterisk indicates a statistically significant difference.
Risk factors for sporadic AD are linked to human behaviour, and include dietary habits, tobacco use, and even environmental pollution.29 Epidemiological studies have shown that individuals with low levels of schooling, history of brain injury, high calorie intake, or sedentary lifestyles are at greatest risk of AD.30 Alterations to higher-order multimodal association areas of the neocortex are responsible for the progressive impairment of cognitive ability, including executive dysfunction (prefrontal cortex) and semantic memory.2,13,15
The physical and functional alterations occurring in AD clearly progress as the disease advances; the locomotor system is also compromised, although to a lesser extent.3 Motor alterations are observed in the first stage of AD, as well as apathy and a tendency to sedentary behaviour, favouring immobility and accelerating patients’ physical decline.6 Posture and gait control begin to show alterations during the second (intermediate) stage of the disease; physical activity is associated with reduced risk of cognitive dysfunction, AD, and dementia in general, protecting memory.31 AD affects sensory and motor regions of the central nervous system; interventions targeting sensorimotor deficits may improve patients’ functional status as the disease progresses.32 Various experiments involving physiotherapy and/or voluntary exercise in transgenic models of AD have shown that exercise and/or environmental enrichment can promote neuronal survival, resistance to brain insult, and cerebral vascularisation; stimulate neurogenesis; improve learning; and contribute to maintaining cognitive function.33 In 3xTg-AD mice, exercise wheels have been shown to represent a unique environmental manipulation and promote neurobehavioural plasticity in terms of gene-environment interactions with relevance to pathogenesis.33
In designing this study, we identified an association between impaired performance in the open field test (behaviour involving the motor cortex) and neuronal alterations, amyloidogenesis, and astrogenesis in the M1 region of 3xTg-AD mice at an intermediate stage (11 months). One of the main neuropathological alterations described in AD is decreased brain volume and weight, which is associated with a decrease in the number of neurons.34 Our findings are consistent with this: we observed significant neuronal alterations in the 3xTg-AD group compared to non-Tg mice, which suggests that M1 neurons in this model are susceptible to damage (cytoplasmatic hyperchromasia, cellular shrinking, and nuclear fragmentation). These findings are comparable to those of other transgenic mouse studies of AD reporting neuronal loss in the hippocampus, which is related to spatial memory, as well as in the cortex.15,34,35 One proposed explanation is synaptopathy, a mechanism of damage to cortical circuits whereby synaptic contacts are lost in cellular networks involved in cognitive function.24 We detected extracellular deposition of Aβ and hyperphosphorylated tau protein, as has been reported by other authors17,35; this was associated with cognitive impairment, confirming that this model presents the human form of the disease. The impaired performance in the open field test also increased its power as an indicator of the motor impairment associated with the presence of the pathological characteristics of both proteins; tau protein appeared to be the main cause of neuronal death in AD. Hippocampal place cell activity is reported to contribute to unstable spatial representation and spatial memory deficits.36
The soluble or monomeric form of the Aβ peptide is neurotoxic and has been shown in several in vitro and in situ studies to trigger an inflammatory reaction in the brains of patients with AD.37 Specifically, microglia produce a series of proinflammatory cytokines and mediators in response to Aβ and activate astrocytes, which become involved in the inflammatory process, creating a “vicious circle” of neuroinflammation.38 Other studies analyse Aβ-related pathways regulating the brain’s innate immunity, with a view to potential therapeutic applications.39 In the brain, reactive astrocytes surround Aβ plaques; this represents an endogenous defence mechanism against plaque deposition. However, the persistent activation of these cells and the associated inflammation may also contribute to AD progression.40 Astrocytes actively communicate with neurons, modulating synaptic plasticity by releasing such gliotransmitters as glutamate and D-serine.38 In AD, astrocytes may undergo morphological and functional changes (astrogliosis), demonstrated by increased GFAP expression22,41 and astrocytic hypertrophy,21,26 as well as such anatomical alterations as increased numbers of astrocytic processes, which may be associated with the increased production of factors potentially harmful to adjacent cells.40 Therefore, neurodegenerative processes of astrocytes may impact the brain’s defence against damage by activating a variety of neuroprotective functions.41,42
Astrogliosis has also been reported in post mortem tissue samples from patients with AD compared to controls, and increases with age.43 Astrocytes therefore constitute a key element in Alzheimer-type dementia development.44 Our findings are consistent with this as we found greater GFAP immunoreactivity in the hippocampus than in the primary motor cortex. It may be the case that this astrocytic hypertrophy increases as the disease progresses, and that microglia and immune cells play a role in the onset and/or progression of the disease. Extracellular aggregation of Aβ peptide in typical neuritic plaques generates a constant inflammatory environment, promoting neuronal damage and blood-brain barrier alterations.44
Therefore, neuronal and glial dysfunction and the pathways regulating inflammation of the brain play an important role in intermediate stages of the disease, when neuroplasticity mechanisms are limited and damage is irreversible. It is important to define the early stages of the disease and establish potential treatment strategies in the periods when it may be possible to stop the degenerative processes responsible for the disease.
FundingThis work was partially funded by CONACYT (grant no. 295523; project no. CB-2012/178841) and UNAM-DGAPA (IN201613).
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors would like to thank the Institute of Neurobiology’s microscopy, proteogenomics, and behaviour units, and laboratory technicians A. Aguilar Vázquez and M. Servín García for their help in caring for the animals.
Please cite this article as: Orta-Salazar E, Feria-Velasco AI, Díaz-Cintra S. Alteraciones en la corteza motora primaria en la enfermedad de Alzheimer: estudio en el modelo 3xTg-AD. Neurología. 2019;34:429–436.
